
Genetics of congenital anomalies of the kidney and urinary tract
Danuta Zwolińska 1 , Dorota Polak-Jonkisz 1 , Irena Makulska 1Streszczenie
Wrodzone wady nerek i układu moczowego (congenital anomalies of the kidney and urinary tract – CAKUT) występują z częstością 1 na 500 żywych urodzeń i są częstą przyczyną niewydolności nerek już w okresie dzieciństwa. Obejmują wiele anomalii nerek, dróg odprowadzających mocz oraz pęcherza i cewki moczowej. Większość z nich ma charakter sporadyczny i izolowany, ale mogą też występować rodzinnie i być częścią zdefiniowanego zespołu klinicznego. Do zrozumienia patogenezy tych wad niezbędna jest znajomość rozwoju nerek i układu moczowego. Proces ten jest bardzo skomplikowany i wymaga precyzyjnej integracji wielu różnorodnych komórek pochodzenia embrionalnego. Embriogeneza układu moczowego kontrolowana jest przez wiele czynników na każdym jej etapie. Autorki skoncentrowały się głównie na czynnikach genetycznych, które prowadzą do zaburzenia procesów morfogenezy, a zwłaszcza do zaburzeń interakcji między pączkiem moczowodowym a mezenchymą metanefronu, co skutkuje powstaniem większości wad typu CAKUT. W pracy przedstawiono również istotne wyniki badań genetycznych przeprowadzonych na modelach zwierzęcych oraz u ludzi z anomaliami typu CAKUT. Do pełnego zrozumienia skomplikowanej molekularnej sieci, której zaburzenie prowadzi do CAKUT, niezbędne są jednak dalsze badania. Ich wyniki będą pomocne przy ustalaniu nowych strategii prewencyjnych i terapeutycznych u pacjentów z wrodzonymi anomaliami nerek i układu moczowego.
Słowa kluczowe:wady wrodzone układu moczowego • podłoże genetyczne wad • embriogeneza układu moczowego • dzieci
Summary
Congenital anomalies of the kidney and urinary tract (CAKUT) occur at a frequency of 1 in 500 live births and are a common cause of renal insufficiency in childhood. CAKUT encompass a wide spectrum of malformations including anomalies of the kidney, collecting system, bladder and urethra. Most cases of CAKUT are sporadic and limited to the urinary tract, but some of them are syndromic or associated with positive family history. To understand the basis of human renal anomalies, knowledge of kidney and urinary tract development is necessary. This process is very complicated, requires precise integration of a variety of progenitor cell populations of diverse embryonic origins and is controlled by many factors at every stage of development. This review focuses on the genetic factors leading to developmental errors of important morphogenetic processes, particularly in metanephric kidney induction and ureteric bud branching. The essential results of genetic studies in regard to CAKUT, performed on experimental models and in humans, are presented. However, further investigations are required to complete understanding of the complex molecular network, which will help us to determine novel preventive and therapeutic strategies for CAKUT.
Key words:congenital anomalies of the kidney and urinary tract • genetics of CAKUT kidney and urinary tract development • children
Wykaz skrótów:
AGTR1, AGTR2 – receptory angiotensyny II typu 1 i 2 (angiotensin receptor 1,2); BMP4 – białko morfogeniczne 4 (bone morphogenetic protein 4); CAKUT – wrodzone wady nerek i układu moczowego (congenital anomalies of the kidney and urinary tract); EGF – naskórkowy czynnik wzrostu (epidermal growth factor); FGF – czynnik wzrostu fibroblastów (fibroblast growth factor); FGFR – receptor czynnika wzrostu fibroblastów (fibroblast growth factor receptor); GDNF – czynnik neurotroficzny pochodzenia glejowego (glial-derived neurotrophic factor), GFRA α1 – receptor a1 czynnika neurotroficznego pochodzenia glejowego (GDNF-family receptor α1); HLA – antygeny zgodności tkankowej (human leukocyte antygens); HNF1β – jądrowy czynnik hepatocytów 1β (hepatocyte nuclear factor 1β); MEN2 – zespół gruczolakowatości wewnątrzwydzielniczej typu 2 (multiple endocrine neoplasia type 2); MM – mezenchyma metanefronu; OPM – odpływ pęcherzowo-moczowodowy; PCHN – przewlekła choroba nerek; PM – pączek moczowodowy; RAS – układ renina-angiotensyna (renin-angiotensin system); RET – receptor kinazy tyrozynowej (REarranged during transfection); SNN – schyłkowa niewydolność nerek.
Wprowadzenie
Wrodzone wady nerek i układu moczowego (CAKUT- congenital anomalies of the kidney and urinary tract) występują z częstością 1 na 500 żywych urodzeń i stanowią poważny problem kliniczny. Według różnych rejestrów światowych, w 30-43% przypadków, są one przyczyną schyłkowej niewydolności nerek (SNN) u dzieci [50], odsetek ten wzrasta do 60%, jeśli weźmiemy pod uwagę wcześniejsze stadia przewlekłej choroby nerek (PCHN) [15]. Z polskiego rejestru dzieci dializowanych w latach 2006-2007 wynika, że CAKUT było pierwotną przyczyną SNN u ponad połowy z nich [63]. Do wad typu CAKUT należą wady nerek (aplazja, hipoplazja, dysplazja, zdwojenia nerek), anomalie dróg odprowadzających mocz (zwężenia moczowodu, moczowód olbrzymi, zdwojenie moczowodów), pęcherza moczowego (torbiel ujścia moczowodu, odpływ pęcherzowo-moczowodowy) oraz cewki moczowej (zastawki cewki tylnej). Większość z tych wad ma charakter sporadyczny i izolowany, ale mogą też występować rodzinnie i być częścią zdefiniowanego zespołu klinicznego. Wskazuje to na istotną rolę zaburzeń genetycznych w patogenezie CAKUT. Na ostateczny fenotyp wpływają czynniki środowiskowe, które nakładając się na defekty genetyczne mogą dawać różnorodne obrazy kliniczne przy tej samej mutacji genowej. Udowodniono to zarówno na modelach zwierzęcych, jak i w badaniach u ludzi [1].
Rozwój układu moczowego w życiu płodowym to proces bardzo skomplikowany, podlegający regulacji wielu genów. Nic więc dziwnego, że wady typu CAKUT należą do najczęstszych anomalii rozwojowych.
Embriogeneza układu moczowego
Układ moczowy powstaje z mezodermy pośredniej. Rozwija się z niej nerka, moczowód i trójkąt pęcherza. Sam pęcherz i cewka moczowa są pochodzenia endodermalnego. W procesie rozwoju powstają i różnicują się kolejno trzy struktury: przednercze (pronephros), śródnercze, zwane inaczej pranerczem (mesonephros) i nerka ostateczna (metanephros). Przednercze powstaje na początku 4 tygodnia po zapłodnieniu, po obu stronach aorty, zbudowane jest z kanalików utworzonych z mezodermy pośredniej okolicy szyjnej. Zazwyczaj występują w liczbie 7-10 par, przy czym w miarę rozwoju kolejnych, starsze ulegają zanikowi. Kanaliki uchodzą do wspólnego przewodu, który w późniejszym okresie przekształca się w przewód pranercza, zwanego kanałem Wolffa. U człowieka przednercze nie jest czynne i zanika już w pierwszej połowie drugiego miesiąca. Śródnercze zaczyna się rozwijać pod koniec 4 tygodnia życia płodowego, ma więcej kanalików, które przybierają kształt esowaty, a ślepy ich koniec tworzy ciałko nerkowe. Drugim końcem kanaliki łączą się z przewodem Wolffa uchodzącym do steku. Do każdego ciałka nerkowego wpukla się pętla naczyniowa tworząc po przekształceniach kłębuszek nerkowy, gdzie odbywa się przesączanie, nieprowadzące jednak do wytwarzania moczu ostatecznego. Pod koniec 8 tygodnia życia płodowego większość kanalików śródnercza zanika. Zaburzenia różnicowania tej struktury mogą prowadzić do agenezji nerek oraz wad gonad i nadnerczy. Nerka ostateczna zaczyna się tworzyć w 5 tygodniu życia płodowego z dwóch struktur: pączka moczowodowego (PM), który jest uwypukleniem kanału Wolffa w części zbliżonej do steku oraz z mezodermy nerki ostatecznej czyli mezenchymy metanefronu (MM). Rozwój nerki rozpoczyna się z chwilą wniknięcia PM do mezodermy, która tworzy zbitą masę tkankową [54]. Szypuła PM przekształca się w moczowód, a bańka wnikająca w mezodermę ulega podziałowi i kolejnym rozgałęzieniom, z nich powstaną: miedniczka, kielichy, przewody brodawkowe i cewki zbiorcze. Rozwój nefronów rozpoczyna się około 8 tygodnia życia płodowego przez skupianie się ognisk mezodermy wokół obu stron bańki pączka moczowodowego. Tworzy się pęcherzyk, zwany sferoidem, który wkrótce przybiera kształt przecinkowatego, a następnie esowatego kanalika Jego poszczególne części przeobrażają się w różne struktury nefronu, co przedstawia ryc. 1. Pęcherz moczowy powstaje między 4-7 tygodniem życia płodowego z zatoki moczowo-płciowej, która została utworzona w wyniku podziału steku przez przegrodę. Do niej uchodzi przewód Wolffa, który przekształca się w moczowód.
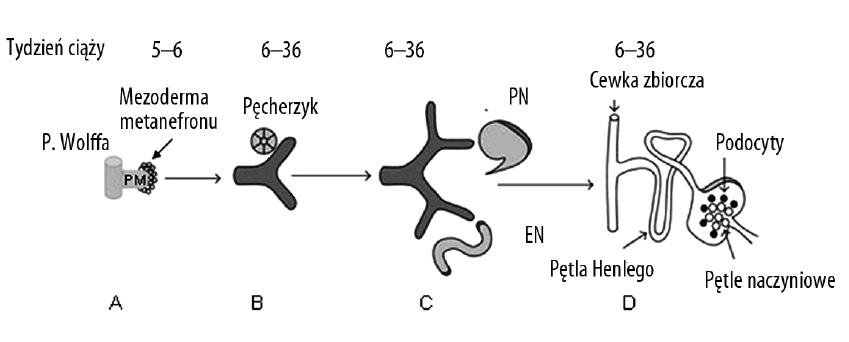
Ryc. 1. Prawidłowy rozwój nerki i układu moczowego; PM – pączek moczowodowy, PN – przecinkowaty kształt przyszłego nefronu, EN – esowaty kształt przyszłego nefronu (wg [50] zmodyfikowano)
Teoria pączka moczowodowego
Czynniki, które wpływają na proces wnikania pączka moczowodowego do mezenchymy metanefronu odgrywają główną rolę w patogenezie CAKUT [39]. Obserwacja kliniczna dotycząca współistnienia hipoplazji/dysplazji nerek z odpływem pęcherzowo-moczowodowym (OPM) zainspirowała powstanie teorii pączka moczowodowego, która pozwala na wyjaśnienie patogenezy wielu wad typu CAKUT. Niektóre z nich przedstawia ryc. 2. Nieprawidłowe umiejscowienie PM (C) sprawia, że trafia on na niewielką ilość zróżnicowanej bądź niezróżnicowanej mezenchymy metanefronu, co prowadzi do rozwoju nerki hipoplastycznej bądź dysplastycznej. Jednak ektopiczny dystalny zawiązek moczowodu łącząc się z proksymalną częścią trójkąta pęcherza z krótkim odcinkiem podśluzówkowym sprzyja OPM. Z kolei powstanie dodatkowego pączka moczowodowego prowadzi do całkowitego zdwojenia moczowodu i nerki, której górna część jest histologicznie niezróżnicowana, dysplastyczna. Dodatkowy zawiązek moczowodu determinuje również nieprawidłowe umiejscowienie ujść moczowodowych w pęcherzu. Zgodnie z prawem Weigerta-Meyera w przypadku takiego zdwojenia, górna część drenowana jest przez moczowód uchodzący w nieprawidłowym miejscu do pęcherza (c), a dolna część nerki przez moczowód uchodzący w miejscu prawidłowym (b). Tym samym, moczowód segmentu górnego, ze względu na krótszy odcinek kanału podśluzówkowego z bocznie położonym ujściem, ma szczególne predyspozycje do wystąpienia odpływu [16].

Ryc. 2. Schemat powstawania zdwojonej nerki i moczowodów. Zgodnie z prawem Weigerta-Meyera i Mackie-Stephensa moczowód uchodzący w prawidłowym położeniu drenuje histologicznie prawidłową dolną część nerki, a w nieprawidłowym położeniu – część górną, dysplastyczną nerki (wg [16] zmodyfikowano)
Czynniki genetyczne biorące udział w patogenezie wad typu CAKUT
Rozwój układu moczowego przebiega pod kontrolą licznych genów. Kodują one białka sygnałowe, czynniki transkrypcyjne, czynniki wzrostu, a także receptory białek biorących udział w morfogenezie. Czynniki te ściśle ze sobą współpracują na poszczególnych etapach, a ich właściwe interakcje warunkują prawidłowy ostateczny kształt i funkcję układu moczowego [43,51,62]. Błędy genetyczne trzech procesów rozwojowych (indukcji pączka moczowodowego, połączenia moczowodowo-pęcherzowego, dojrzewania mechanizmu warunkującego odpowiednią perystaltykę miedniczkowo-moczowodową) są odpowiedzialne za powstawanie wad typu CAKUT [48]. Najczęstszą i najliczniejszą grupę stanowią anomalie, które są związane z defektami genów kontrolujących indukcję pączka moczowodowego [13,31,59].
Molekularna regulacja indukcji pączka moczowodowego i powstawania wczesnych rozgałęzień
Do najważniejszych czynników kontrolujących wczesne stadium morfogenezy należy GDNF (glial-derived neurotrophic factor), którego ekspresja w mezenchymie metanefronu jest bardzo wysoka. Po przyłączeniu się do swoistego receptora GFRA α1 (GDNF-family receptor α1) aktywuje on receptor kinazy tyrozynowej RET (REarranged during transfection) inicjując proces wzrostu pączka moczowodowego w kierunku mezenchymy metanefronu oraz uruchamiając kaskadę białek sygnałowych prowadzących do proliferacji komórek epitelialnych i tworzenia rozgałęzień. Ekspresja obu receptorów jest wyraźna w przewodzie Wolffa.
Aby proces indukcji pączka moczowodowego przebiegał prawidłowo i zakończył się powstaniem jednego, umiejscowionego właściwie w moczowodzie, oś GDNF-RET jest ściśle kontrolowana przez wiele genów, zarówno o działaniu stymulującym jak i hamującym. Stymulujące działanie mają przede wszystkim geny trzech czynników transkrypcyjnych, których ekspresja w mezodermie pośredniej jest wyraźnie zaznaczona już w bardzo wczesnych fazach rozwoju. Są to geny: Paired-box 2 (PAX 2), Eyes-absent homolog 1 (EYA1) i Sine-oculis homeobox homolog 1(SIX1), które w molekularnej kaskadzie uaktywniają bezpośrednio GDNF [9]. Na modelach zwierzęcych wykazano, że u pozbawionych tych genów myszy, wskutek braku wytworzenia się pączka moczowodowego dochodzi do agenezji nerek [39]. Do innych molekularnych aktywatorów GDNF należą geny: HOXA11, HOXC11, HOXD11, LIM 1 i EMX2, których ekspresja jest widoczna w przewodzie Wolffa. Ich całkowity brak wywołuje u zwierząt doświadczalnych podobny efekt fenotypowy [35,36].
Negatywne działanie na oś GDNF-RET mają natomiast geny: Forkhead box protein1c (FOXC1) oraz Slit homolog 2 (SLIT2) i jego receptor Rounabout homolog 2 (ROBO2). Zaburzenia w tych genach u myszy mogą prowadzić do nadliczbowych i ektopicznych moczowodów. Podobny efekt wywołują mutacje genów kodujących białko morfogeniczne 4,7 (bone morphogenetic protein 4 – BMP4, BMP7 oraz Sprouty 1 (SPRY1). Ekspresja tych genów jest najwyższa w MM, w pobliżu przewodu Wolffa [12,34,42]. Szczególna rola w prawidłowym tworzeniu moczowodu i jego połączenia z nerką przypada białku BMP4. Odpowiada ono bowiem za prawidłową nawigację pączka moczowodowego [16,28]. Pączek moczowodowy może się połączyć z mezenchymą metanefronu tylko w miejscu, gdzie brak jest białka BMP4, ale wydzielany jest czynnik wzrostowy GDNF. Jeśli proces ten zostanie zaburzony, pączek moczowodowy może wykształcić się w innym miejscu lub też w kilku miejscach, prowadząc m.in. do zdwojenia i dysplazji nerek oraz OPM [29]. Ten sam efekt można uzyskać w przypadku zaburzeń genetycznych dotyczących białka gremliny, która wpływa hamująco na BMP4 lub przy defektach innych genów, pośrednio powiązanych z osią GDNF-RET [10,27].
Ważną rolę na wielu etapach rozwoju układu moczowego odgrywają czynniki wzrostu fibroblastów (FGF – fibroblast growth factors), zwłaszcza: 2,7,8, i 10 oraz ich receptory (FGFR – fibroblast growth factor receptors) [10]. Wpływają one nie tylko na wzrost i dojrzewanie mezenchymy metanefronu i pączka moczowodowego, ale także na proces rozgałęziania i formowania ostatecznych nefronów. Nic więc dziwnego, że w modelach mysich, zwłaszcza z defektem receptora FGFR2 wykazano rozmaite nerkowe fenotypy: agenezję i hipoplazję nerek, zdwojenie moczowodów i nerek, a także OPM [14]. Opisywane są także mutacje genów FGFR, odpowiedzialnych u ludzi za rozwój zespołów klinicznych, w skład których wchodzą różne nieprawidłowości w układzie moczowym. Na przykład w zespole Kallmana z defektem genu FGFR1 może występować agenezja nerki, w zespole Pfeiffera z mutacją genu FGFR2 obserwować można wysokiego stopnia OPM, a w zespole ciężkiej dysplazji szkieletowej, którego przyczyną jest nieprawidłowy gen FGFR3 – hipoplazję lub dysplazję wielotorbielowatą nerek [3]. Układ renina, angiotensyna (renin-angiotensin system – RAS), istotny regulator kontroli ciśnienia tętniczego i gospodarki wodno-elektrolitowej, jest również ważnym czynnikiem biorącym udział w morfogenezie układu moczowego. Ekspresja wszystkich jego składowych, a zwłaszcza receptorów angiotensyny II typu 1 i 2 (AGTR1, AGTR2 – angiotensin receptor 1, 2) jest widoczna w okresie embriogenezy już od 5 tygodnia po zapłodnieniu. W badaniach eksperymentalnych wykazano, że defekty tych genów prowadzą do rozmaitych anomalii typu CAKUT [33,41]. Stwierdzono m.in. olbrzymie wodonercze z rdzeniową dysplazją nerek u myszy pozbawionych genu kodującego ATGR2. Inaktywacja tego genu może prowadzić również do ektopii nerek, ich zdwojenia, zwężenia podmiedniczkowego i przypęcherzowego moczowodów, dysplazji i hipoplazji nerek, nerki wielotorbielowatej oraz OPM [61]. Taka różnorodność fenotypowa jest związana z różnymi miejscami działania składowych RAS w okresie embriogenezy. Z jednej strony wpływają one bezpośrednio na mięśniówkę gładką moczowodów, co prowadzi do zaburzeń ich perystaltyki, z drugiej zaś, poprzez interakcję z innymi czynnikami, modyfikują przebieg poszczególnych etapów rozwojowych układu moczowego. Wpływając na PAX2, GDNF, RET, BMP4 oraz na receptor naskórkowego czynnika wzrostu (EGF- epidermal growth factor) regulują proces indukcji pączka moczowodowego. W ostatnich latach wykazano, że na powstawanie anomalii w dolnych drogach moczowych mają wpływ geny kodujące uroplakiny. Są to glikoproteiny występujące w błonie komórek urotelialnych od strony światła dróg moczowych. Ich rolą jest przede wszystkim uszczelnienie urotelium, zmniejszenie jego przepuszczalności dla jonów i substancji rozpuszczonych w moczu. Naruszenie integralności tego nabłonka może skutkować rozwojem ciężkich OPM i wodonercza, co wykazano w badaniach eksperymentalnych [60].
Fenotyp osobniczy a zaburzenia genów regulujących morfogenezę układu moczowego w modelach mysich
Przytoczone wyżej dane wskazują jak bardzo skomplikowana jest regulacja procesu embriogenezy. Wzajemna interakcja tak wielu czynników biorących w nim udział jest przyczyną powstawania różnych fenotypów przy mutacji tego samego genu lub jednego fenotypu przy mutacjach różnych genów. Wykazały to liczne badania w modelach mysich, pozbawionych badanych genów lub zawierających geny zmutowane. Ich przykłady podano w tabeli 1.
Tabela 1. Mutacje genowe a fenotyp nerkowy

Jak wiele odmian może mieć defekt tego samego genu przekonać się można na przykładzie genu RET. Fizjologicznie odgrywa istotną rolę w wielu punktach morfogenezy poprzez trzy różne szlaki sygnałowe. Uczestniczy w indukcji PM, wpływa na proces kształtowania się rozgałęzień, gwarantuje rozwój pojedynczego pączka moczowodowego oraz wzrost i dojrzewanie moczowodu. W zależności od miejsca uszkodzenia genu RET dochodzi do różnorodnych wad. Może to być: jedno- lub obustronna agenezja, agenezja z hipoplazją, obustronna hipoplazja, obustronny moczowód olbrzymi z hipodysplazją, a także moczowód olbrzymi jednostronny. W tym miejscu trzeba podkreślić, że ten receptor kinazy tyrozynowej, poprzez swoje działanie proonkogenne, może także wpływać na zaburzenia w innych narządach. Mutacje inaktywujące genu RET prowadzić mogą do rozwoju choroby Hirschprunga, zaś mutacje aktywujące do zespołu gruczolakowatości wewnątrzwydzielniczej typu 2 (MEN-2 – multiple endocrine neoplasia type 2) i nowotworów tarczycy [17,38,44].
Podobna sytuacja odnosi się do genu kodującego białko BMP4. Myszy z mutacją tego genu mogą prezentować różne obrazy fenotypowe: hipodysplazja nerek, wodonercze z olbrzymimi moczowodami czy zdwojony moczowód i nerki [8]. Innym przykładem jest defekt genetyczny FGF i jego receptorów, który w modelach doświadczalnych generował różnorodne wady typu CAKUT: hipodysplazja nerek, zdwojenie nerek i moczowodów, zwężenie moczowodów z wodonerczem czy OPM [41]. Dodatkowo trzeba podkreślić, że na ostateczny fenotyp wady wpływa siła ekspresji zmodyfikowanych genów.
Zaburzenia genetyczne w wadach typu CAKUT w badaniach klinicznych
Przybywa badań dotyczących zaburzeń genetycznych w wadach typu CAKUT u ludzi. Ich wyniki, w większości przypadków, mają na razie znaczenie poznawcze, jednak w poszczególnych sytuacjach już teraz mogą być pomocne w praktyce klinicznej.
Hipodysplazja nerek
Ponieważ hipodysplazja nerek u dzieci ma swój znaczący udział w rozwoju SNN (20%), wiele badań genetycznych przeprowadzono właśnie w tej grupie chorych [23,61]. Za znaczącym udziałem czynników genetycznych w patogenezie tej wady przemawiało to, iż występuje ona nie tylko jako wada izolowana, ale również jako składowa zespołów dziedziczących się autosomalnie dominująco lub recesywnie [1,22,46,53]. Są one związane najczęściej z mutacjami następujących genów:
PAX2:renal-coloboma syndrome (OPM, dysplazja nerwu wzrokowego, szczelina oka, rzadziej niedosłuch) [45].
HNF1b: hepatocyte nuclear factor 1b (jądrowy czynnik 1b ηepatocytów) – renal cysts and diabetes syndrome (torbielowatość nerek, cukrzyca typu MODY, zaburzenia wątrobowe) [25,32].
EYA1 i SIX1: branchio-oto-renal syndrome (przetoka oskrzelowa, dołki przeduszne, zniekształcenia małżowiny, zaburzenia słuchu) [21,37].
SALL1: Townes-Brocks syndrome (OPM, zastawka cewki tylnej, zaburzenia w układach: szkieletowym, nerwowym, sercowym, zaburzenia wątrobowe). W tabeli 2 przedstawiono również inne zespoły chorobowe uwarunkowane genetycznie i związane z hipodysplazją nerek [11,40].
Tabela 2. Genetycznie uwarunkowane zespoły z towarzyszącą hipodysplazją u ludzi

Jedno z większych opracowań, przeprowadzonych w ramach dużego badania ESCAPE, objęło 100 dzieci z hipodysplazją nerek i PCHN w stadium 2-4, z 99 niespokrewnionych rodzin. W jednej rodzinie dwoje dzieci ujawniało zmiany nerkowe. Z badania wykluczono chorych z wadami pęcherza, zastawką cewki tylnej oraz pacjentów z zespołami suszonej śliwki i Bardeta-Biedla. W 62 przypadkach ujawniono dodatkowo inne wady, głównie OPM, a u 12% dzieci stwierdzono dodatni nerkowy wywiad rodzinny. Badano geny – PAX2, EYA1, SIX1, SALL1, HNF1β, a więc geny, których mutacje odpowiadają za przedstawione wcześniej zespoły. Przeprowadzono również dokładne badania w kierunku objawów pozanerkowych u dzieci oraz członków ich rodzin w przypadku wykrycia defektów w wyżej wymienionych genach. W sumie mutacje PAX2 stwierdzono u 7 dzieci. U 5 z nich, oraz u 2 rodziców wykryto objawy pozanerkowe, typowe dla zespołu renal-coloboma. Podkreślić należy, że stwierdzono je dopiero po identyfikacji zaburzeń genetycznych. W zakresie genu HNF1β wykryto mutacje u 8 chorych, w tym 1 de novo. Co ciekawsze, aż u 6 z nich stwierdzono zmiany torbielowate, a u 3 dzieci oraz u 3 członków ich rodzin objawy pozanerkowe, typowe dla zespołu renal cysts and diabetes. W pojedynczych przypadkach wykazano mutacje w pozostałych genach; u pacjenta z mutacją EYA1 obserwowano kliniczne cechy zespołu branchio-oto-renal, których z kolei nie stwierdzono u 2 dzieci z mutacją genu SIX1. Wyciągnięte z pracy wnioski są ważne z klinicznego punktu widzenia. Wskazują bowiem nie tylko na częstą asocjację genów HNF1β i PAX2 z hipodysplazją nerek w porównaniu do genów EYA1 i SIX1, ale również na konieczność przeprowadzenia dokładnych badań pod kątem dyskretnych objawów pozanerkowych u dzieci, u których stwierdzono wyżej wymienione defekty genowe [57].
W kolejnej pracy tego samego zespołu badano po raz pierwszy mutacje genów: SIX 2 i BMP4 u dzieci z hipodysplazją nerek. W dużej grupie, obejmującej 250 pacjentów, wykryto je u 10 dzieci, u 5 w genie SIX2 i u 5 – w genie BMP4. Warto podkreślić, że u połowy z tych dzieci stwierdzono dodatkową wadę w postaci OPM, co potwierdza obserwacje przeprowadzone na modelach zwierzęcych o możliwości występowania różnych fenotypów przy defekcie tego samego genu [58]. Określając poziom mRNA w 3 wariantach genu BMP4 udało się również udowodnić czynnościowy związek defektu BMP4 z wadami typu CAKUT. Stwierdzono bowiem, zwłaszcza w zakresie jednego z wariantów, istotnie niższą jego wartość, co wskazuje na mniejszą skuteczność transkrypcyjną, a w rezultacie na silniejszy efekt hamujący zmutowanego genu. Dodatkowo wykazano, że ekspresja i umiejscowienie BMP4, zarówno w siateczce endoplazmatycznej jak i w lizosomach, są nieprawidłowe [52]. W ramach badania ESCAPE analizowano również mutacje genu kodującego uroplakinę III u dzieci z hipodysplazją nerek, stwierdzając, że występują one rzadko w tej grupie chorych [19].
Interesujące są wyniki badań, których celem było określenie fenotypu nerkowego związanego z mutacją genu HNF1β, a więc genu odpowiedzialnego za zespół, w którym oprócz cukrzycy stwierdza się obecność torbieli w nerkach. W grupie 80 dzieci z różnymi wadami, w tym z wielotorbielowatością i pojedynczymi cystami w nerkach, mutacje tego genu stwierdzono aż u 25 osób. Występowały one istotnie częściej u dzieci z obecnością torbieli (p<0,001) w porównaniu do pacjentów z hipodysplazją nerek czy agenezją. Nie wykazano natomiast korelacji zmutowanych genów HNF1β z przesączaniem kłębuszkowym [56].
Agenezja nerek a RET
Niewiele jest danych dotyczących zaburzeń genetycznych RET. Pierwsze badania przeprowadzono na martwych płodach z jedno- bądź obustronną agenezją lub ciężką dysplazją nerek. Obecność mutacji RET wykryto u 9 na 33 płody, częściej przy obustronnym braku nerek, co przy wyjątkowym występowaniu mutacji tego genu w populacji ogólnej, wskazywało na istotny związek przyczynowo-skutkowy. W tej samej pracy badano także geny GNDF i GFRA1 nie wykazując takiej zależności [49]. Powyższej obserwacji nie potwierdziły jednak najnowsze badania, przeprowadzone na 105 martwych płodach. Wykazano jedynie nieco częstsze występowanie jednego z wariantów RET w porównaniu z ogólną populacją (6 vs. 2%, p<0,01). Nie stwierdzono natomiast, podobnie jak w poprzedniej pracy, pozanerkowych anomalii, które mogą wystąpić w przypadku mutacji RET [18].
Odpływ pęcherzowo-moczowodowy
Odpływ pęcherzowo-moczowodowy jest jedną z częstszych wad układu moczowego i dotyczy prawie 1% populacji ogólnej [7]. W 30-50% występuje u członków tej samej rodziny, a w 100% obserwuje się go u monozygotycznych bliźniąt. Te kliniczne obserwacje od ponad 30 lat stały się podstawą do poszukiwania genetycznego podłoża wady. Wcześniejsze badania wykazały powiązanie OPM z układem HLA, w szczególności z antygenami HLA-B12 u kobiet oraz HLA-A9 w połączeniu z HLA-BW15 u mężczyzn [55]. Z kolei grupa japońska wskazywała na związek HLA-DR11 i niektórych alleli HLA-DRB1 u dzieci [20]. W ostatnich 20 latach prowadzone są badania genomu ludzkiego oraz badania poszukujące defektów w poszczególnych genach. Liczba prac, które potwierdzają genetyczne tło OPM jest bardzo duża. Nie ulega wątpliwości, że odpływy pęcherzowo-moczowodowe są genetycznie heterogenną grupą wad. Rodzinnie OPM może występować z różnym modelem dziedziczenia, jako cecha autosomalna dominująca o różnej penetracji i ekspresji genu, jako cecha autosomalna recesywna lub uwarunkowana wieloczynnikowo [4]. Opisano również rodzinę, w której OPM był dziedziczony z chromosomem X. Dotąd zidentyfikowano wiele genów kandydatów, które mogą odgrywać rolę w patogenezie OPM. Są to między innymi geny kodujące RAS, SLIT2 i jego receptor ROBO2, geny kodujące uroplakiny, TGF-β, a także EYA1, PAX2, SALL1, LIM-1, FGFR2, RET, SIX1, VEGF [6,14,19,30]. Lista genów jest znacznie dłuższa i wciąż otwarta. Trzeba podkreślić, że obecnie wiele badań w odniesieniu do mutacji tego samego genu przynosi sprzeczne wyniki. Świadczy to nie tylko o zmienności w obrębie danej populacji, ale prawdopodobnie ma również związek z interakcją innych czynników.
Wpływ czynników środowiskowych na powstawanie wad typu CAKUT
Wśród czynników środowiskowych wymienić należy: zakażenia wirusowe i bakteryjne, promieniowanie rentgenowskie, narkotyki, palenie tytoniu, nadużywanie alkoholu, niedobory żywieniowe oraz leki. Przykładem mogą być inhibitory enzymu konwertującego angiotensynę oraz blokery receptora angiotensyny II. Ekspozycja na te leki w okresie prenatalnym może prowadzić do przerostu aparatu przykłębuszkowego, nadmiernego włóknienia rdzenia nerki oraz zaburzeń lub całkowitego braku różnicowania cewek nerkowych [2,5,24,47]. Skutkiem niedoboru witaminy A może być hipoplazja nerek i rożne anomalie dróg odprowadzających mocz, co wykazano na modelach zwierzęcych [24,26].
Podsumowanie
Za prawidłowy rozwój układu moczowego odpowiada wiele genów kodujących czynniki wzrostu, czynniki transkrypcyjne, swoiste receptory i białka sygnałowe, które ściśle ze sobą współdziałają, a ich mutacje są przyczyną wad typu CAKUT. Mutacje tego samego genu mogą prezentować rożne fenotypy, a różnych genów – podobne fenotypy, co wykazano w licznych badaniach eksperymentalnych. Ponadto ekspresja zmodyfikowanych genów może być bardzo zróżnicowana, co skutkuje różnymi klinicznymi objawami w obecności tego samego defektu. Wady typu CAKUT mogą wystąpić w postaci izolowanej lub w postaci zespołu wad, co przemawia za dodatkową funkcją regulatorową poszczególnych genów, najczęściej kodujących czynniki transkrypcyjne. Choć obecnie wiedza na temat genetycznego podłoża wad typu CAKUT ma głównie znaczenie poznawcze, to w szczególnych sytuacjach może być już teraz pomocna w podjęciu decyzji o rozszerzeniu diagnostyki w kierunku możliwych innych powikłań, nie tylko u pacjentów z wadą, ale także u członków ich rodzin. Precyzyjna diagnostyka genetyczna powinna też spełniać rolę identyfikatora grup dzieci do długofalowej obserwacji.
PIŚMIENNICTWO
[1] Adalat S., Bockenhauer D., Ledermann S.E., Hennekam R.C., Woolf A.S.: Renal malformations associated with mutations of developmental genes: messages from the clinic. Pediatr. Nephrol., 2010; 25: 2247-2255
[PubMed] [Full Text HTML] [Full Text PDF]
[2] Barr M.Jr, Cohen M.M.Jr: ACE inhibitor fetopathy and hypocalvaria: the kidney-skull connection. Teratology, 1991; 44: 485-495
[PubMed]
[3] Bates C.M.: Role of fibroblast growth factor receptor signaling in kidney development. Pediatr. Nephrol., 2011; 26: 1373-1379
[PubMed]
[4] Bertoli-Avella A.M., Conte M.L., Punzo F., de Graaf B.M., Lama G., La Manna A., Polito C., Grassia C., Nobili B., Rambaldi P.F., Oostra B.A., Perrotta S.: ROBO2 gene variants are associated with familial vesicoureteral reflux. J. Am. Soc. Nephrol., 2008; 19: 825-831
[PubMed] [Full Text HTML] [Full Text PDF]
[5] Bos-Thompson M.A., Hillaire-Buys D., Muller F., Dechaud H., Mazurier E., Boulot P., Morin D.: Fetal toxic effects of angiotensin II receptor antagonists: case report and follow-up after birth. Ann. Pharmacother., 2005; 38: 157-161
[PubMed] [Full Text HTML] [Full Text PDF]
[6] Boualia S.K., Gaitan Y., Murawski I., Nadon R., Gupta I.R., Bouchard M.: Vesicoureteral reflux and other urinary tract malformations in mice compound heterozygous for Pax2 and Emx2. PLoS One, 2011; e21529
[PubMed] [Full Text HTML] [Full Text PDF]
[7] Brodkiewicz A., Żakowska A., Bińczak-Kuleta A., Peregud-Pogorzelski J., Ciechanowicz A.: Wsteczny odpływ pęcherzowo-moczowodowy – wszystko zależy od genów? Ann. Acad. Med. Stetin., 2008; 54: 17-23
[PubMed]
[8] Chen F.: Genetic and developmental basis for urinary tract obstruction. Pediatr. Nephrol., 2009; 24: 1621-1632
[PubMed]
[9] Dressler G.R.: Pattering and early cell lineage decision in the developing kidney: the role of PAX genes. Pediatr. Nephrol., 2011; 26: 1387-1394
[PubMed]
[10] Dressler G.R.: Advances in early kidney specification, development and patterning. Development, 2009; 136: 3863-3874
[PubMed] [Full Text HTML] [Full Text PDF]
[11] Faguer S., Pillet A., Chassaing N., Merhenberger M., Bernadet-Monrozies P., Guitard J., Chauveau D.: Nephropathy in Townes-Brocks syndrome (SALL1 mutation): imaging and pathological findings in adulthood. Nephrol. Dial. Transplant., 2009; 24: 1341-1345
[PubMed] [Full Text HTML] [Full Text PDF]
[12] Gonçalves A., Zeller R.: Genetic analysis reveals an unexpected role of BMP7 in initiation of ureteric bud outgrowth in mouse embryos. PLoS One, 2011; 28: e19370
[PubMed] [Full Text HTML] [Full Text PDF]
[13] Hahn H.: Genetics of kidney development: pathogenesis of renal anomalies. Korean J. Pediatr., 2010; 53: 729-734
[PubMed] [Full Text HTML] [Full Text PDF]
[14] Hains D.S., Sims-Lucas S., Carpenter A., Saha M., Murawski I., Kish K., Gupta I., McHugh K., Bates C.M.: High incidence of vesicoureteral reflux in mice with Fgfr2 deletion in kidney mesenchyma. J. Urol., 2010; 183: 2077-2084
[PubMed]
[15] Harambat J., van Stralen K.J., Kim J.J., Tizard E.J.: Epidemiology of chronic kidney disease in children. Pediatr Nephrol., 2011 (w druku)
[PubMed]
[16] Ichikawa I., Kuwayama F., Pope J.C.4th, Stephens F.D., Miyazaki Y.: Paradigm shift from classic anatomic theories to contemporary cell biological views of CAKUT. Kidney Int., 2002; 61: 889-898
[PubMed] [Full Text HTML] [Full Text PDF]
[17] Jain S.: The many faces of RET dysfunction in kidney. Organogenesis, 2009; 5: 177-190
[PubMed] [Full Text HTML] [Full Text PDF]
[18] Jeanpierre C., Mace G., Parisot M., Moriniere V., Pawtowsky A., Benabou M., Martinovic J., Amiel J., Attié-Bitach T., Delezoide A.L., Loget P., Blanchet P., Gaillard D., Gonzales M., Carpentier W., Nitschke P., Tores F., Heidet L., Antignac C., Salomon R., Société Française de Foetopathologie: RET and GDNF mutations are rare in fetuses with renal agenesis or other severe kidney development defects. J. Med. Genet., 2011; 48: 497-504
[PubMed]
[19] Jenkins D., Bitner-Glindzicz M., Malcolm S., Allison J., de Bruyn R., Flanagan S., Thomas D.F, Belk R.A., Feather S.A., Bingham C., Southgate J., Woolf A.S.: Mutation analyses of Uroplakin II in children with renal tract malformations. Nephrol. Dial. Transplant., 2006; 21: 3415-3421
[PubMed] [Full Text HTML] [Full Text PDF]
[20] Kawauchi A., Takahara S., Sada M., Goto R., Nakatani T., Miki T.: Susceptibility to vesicoureteral reflux in Japanese is linked to HLA-DR antigen. Urology, 2001, 58, 1036-1040
[PubMed]
[21] Kimberling W.J., Borsa N., Smith R.J.: Hearing loss disorders associated with renal disease. Adv. Otorhinolaryngol., 2011; 70: 75-83
[PubMed]
[22] Krzemień G., Roszkowska-Blaim M., Kostro I., Wojnar J., Karpińska M., Sekowska R.: Urological anomalies in children with renal agenesis or multicystic dysplastic kidney. J. Appl. Genet., 2006; 47: 171-176
[PubMed]
[23] La Scola C., Hewitt I., Pasini A., Pugliese F., Montini G.: Postnatal management of congenital bilateral renal hypodysplasia. J. Matern. Fetal Neonatal. Med., 2010; 23: Suppl. 3, 97-100
[PubMed]
[24] Martinovic J., Benachi A., Laurent N., Daikha-Dahmane F., Gubler M.C.: Fetal toxic effects and angiotensin-II-receptor antagonists. Lancet, 2001, 358, 241-242
[PubMed]
[25] Martinovic-Bouriel J., Benachi A., Bonniere M., Brahimi N., Esculpavit C., Morichon N., Vekemans M., Antignac C., Salomon R., Encha-Razavi F., Attié-Bitach T., Gubler M.C.: PAX2 mutations in fetal renal hypodysplasia. Am. J. Med. Genet. A, 2010; 152: 830-835
[PubMed]
[26] Merlet-Bénichou C.: Influence of fetal environment on kidney development. Int. J. Dev. Biol., 1999; 43: 453- 456
[PubMed] [Full Text PDF]
[27] Michos O., Goncalves A., Lopez-Rios J., Tiecke E., Naillat F., Beier K., Galli A., Vainio S., Zeller R.: Reduction of BMP4 activity by gremlin 1 enables ureteric bud outgrowth and GDNF/WNT11 feedback signaling during kidney branching morphogenesis. Development, 2007; 134: 2397-2405
[PubMed] [Full Text HTML] [Full Text PDF]
[28] Miyazaki Y., Oshima K., Fogo A., Hogan B.L., Ichikawa I.: Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J. Clin. Invest., 2000; 105: 863-873
[PubMed] [Full Text HTML] [Full Text PDF]
[29] Miyazaki Y., Oshima K., Fogo A., Ichikawa I.: Evidence that bone morphogenic protein4 has multiple biological functions during kidney and urinary tract development. Kidney Int., 2003; 63: 835-844
[PubMed]
[30] Murer L., Benetti E., Artifoni L.: Embryology and genetics of primary vesico-ureteric reflux and associated renal dysplasia. Pediatr. Nephrol., 2007; 22: 788-797
[PubMed]
[31] Nakanishi K., Yoshikawa N.: Genetic disorders of human congenital anomalies of the kidney and urinary tract (CAKUT). Pediatr Int., 2003; 45: 610-616
[PubMed]
[32] Nakayama M., Nozu K., Goto Y., Kamei K., Ito S., Sato H., Emi M., Nakanishi K., Tsuchiya S., Iijima K.: HNF1B alterations associated with congenital anomalies of the kidney and urinary tract. Pediatr. Nephrol., 2010; 25: 1073-1079
[PubMed]
[33] Niimura F., Kon V., Ichikawa I.: The renin-angiotensin system in the development of the congenital anomalies of the kidney and urinary tract. Curr. Opin. Pediatr., 2006; 18: 161-166
[PubMed]
[34] Oxburgh L., Brown A.C., Fetting J., Hill B.: BMP signaling in the nephron progenitor niche. Pediatr. Nephrol., 2011; 26: 1491-1497
[PubMed]
[35] Patterson L.T., Pembaur M., Potter S.S.: Hoxa11 and Hoxd11 regulate branching morphogenesis of the ureteric bud in the developing kidney. Development, 2001; 128: 2153-2161
[PubMed] [Full Text HTML] [Full Text PDF]
[36] Pederson A., Skjong C., Shawlot W.: Lim 1 is required for nephric duct extension and ureteric bud morphogenesis. Dev. Biol., 2005; 288: 571-581
[PubMed]
[37] Pierides A.M., Athanasiou Y., Demetriou K., Koptides M., Deltas C.C.: A family with the branchio-oto-renal syndrome: clinical and genetic correlations. Nephrol. Dial. Transplant., 2002; 17: 1014-1018
[PubMed] [Full Text HTML] [Full Text PDF]
[38] Pini Prato A., Musso M., Ceccherini I., Mattioli G., Giunta C., Ghiggeri G.M., Jasonni V.: Hirschsprung disease and congenital anomalies of the kidney and urinary tract (CAKUT): a novel syndromic association. Medicine (Baltimore), 2009; 88: 83-90
[PubMed]
[39] Pope J.C.4th, Brock J.W.3rd, Adams M.C., Stephens F.D., Ichikawa I.: How they begin and how they end: classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J. Am. Soc. Nephrol., 1999; 10, 2018-2028
[PubMed] [Full Text HTML] [Full Text PDF]
[40] Reardon W., Casserly L.F., Birkenhäger R., Kohlhase J.: Kidney failure in Townes-Brocks syndrome: an under recognized phenomenon? Am. J. Med. Genet. A, 2007; 143A: 2588-2591
[PubMed]
[41] Rigoli L., Chimenz R., di Bella C., Cavallaro E., Caruso R., Briuglia S., Fede C., Salpietro C.D.: Angiotensin-converting enzyme and angiotensin type 2 receptor gene genotype distributions in Italian children with congenital uropathies. Pediatr. Res., 2004; 56: 988-993
[PubMed]
[42] Rozen E.J., Schmidt H., Dolcet X., Basson M.A., Jain S., Encinas M.: Loss of Sprouty1 rescues renal agenesis caused by Ret mutation. J. Am. Soc. Nephrol., 2009; 20: 255-259
[PubMed] [Full Text HTML] [Full Text PDF]
[43] Rumballe B., Georgas K., Wilkinson L., Little M.: Molecular anatomy of the kidney: what have we learned from gene expression and functional genomics? Pediatr. Nephrol., 2010; 25: 1005-1016
[PubMed] [Full Text HTML] [Full Text PDF]
[44] Sampson M.G., Coughlin C.R.2nd, Kaplan P., Conlin L.K., Meyers K.E., Zackai E.H., Spinner N.B., Copelovitch L.: Evidence for a recurrent microdeletion at chromosome 16p11.2 associated with congenital anomalies of the kidney and urinary tract (CAKUT) and Hirschsprung disease. Am. J. Med. Genet A, 2010; 152A: 2618-2622
[PubMed]
[45] Schimmenti L.A.: Renal coloboma syndrome. Eur. J. Hum. Genet., 2011; 19: 1207-1212
[PubMed]
[46] Schreuder M.F., Westland R., van Wijk J.A.: Unilateral multicystic dysplastic kidney: a meta-analysis of observational studies on the incidence, associated urinary tract malformations and the contralateral kidney. Nephrol. Dial. Transplant., 2009; 24: 1810-1818
[PubMed] [Full Text HTML] [Full Text PDF]
[47] Sekine T., Miura K., Takahashi K., Igarashi T.: Children’s toxicology from bench to bed-Drug induced renal injury (1):The toxic effects of ARB/ACEI on fetal kidney development. J. Toxicol. Sci., 2009; 34, Suppl. 2: SP245-SP250
[PubMed] [Full Text PDF]
[48] Shah M.M., Sakurai H., Sweeney D.E., Gallegos T.F., Bush K.T., Esko J.D., Nigam S.K.: Hs2st mediated kidney mesenchyme induction regulates early ureteric bud branching. Dev. Biol., 2010; 339: 354-365
[PubMed] [Full Text HTML] [Full Text PDF]
[49] Skinner M.A., Safford S.D., Reeves J.G., Jackson M.E., Freemerman A.J.: Renal aplasia in humans is associated with RET mutations. Am. J. Hum. Genet., 2008; 82: 344-351
[PubMed] [Full Text HTML] [Full Text PDF]
[50] Song R., Yosipiv I.V.: Genetics of congenital anomalies of the kidney and urinary tract. Pediatr. Nephrol., 2011; 26: 353-364
[PubMed]
[51] Stewart K., Bouchard M.: Kidney and urinary tract development: an apoptotic balancing act. Pediatr. Nephrol., 2011; 26: 1419-1425
[PubMed]
[52] Tabatabaeifar M., Schlingmann K.P., Litwin M., Emre S., Bakkaloglu A., Mehls O., Antignac C., Schaefer F., Weber S.; ESCAPE Trial Group.: Functional analysis of BMP4 mutations identified in pediatric CAKUT patients. Pediatr. Nephrol., 2009; 24: 2361-2368
[PubMed]
[53] Thomas R., Sanna-Cherchi S., Warady B.A., Furth S.L., Kaskel F.J., Gharavi A.G.: HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKD cohort. Pediatr. Nephrol., 2011; 26: 897-890
[PubMed]
[54] Toka H.R., Toka O., Hariri A., Nguyen H.T.: Congenital anomalies of kidney and urinary tract. Semin. Nephrol., 2010; 30: 374-386
[PubMed]
[55] Torres V.E., Moore S.B., Kurtz S.B., Offord K.P., Kelalis P.P.: In search of marker for genetic susceptibility to reflux nephropathy. Clin. Nephrol., 1980; 14: 217-222
[PubMed]
[56] Ulinski T., Lescure S., Beaufils S., Guigonis V., Decramer S., Morin D., Clauin S., Deschenes G., Bouissou F., Bensman A., Bellanné-Chantelot C.: Renal phenotypes related to hepatocyte nuclear factor-1 (TCF2) mutations in a pediatric cohort. J. Am. Soc. Nephrol., 2006; 17: 497-503
[PubMed] [Full Text HTML] [Full Text PDF]
[57] Weber S., Moriniere V., Knuppel T., Charbit M., Dusek J., Ghiggeri G.M., Jankauskiené A., Mir S., Montini G., Peco-Antic A., Wühl E., Zurowska A.M., Mehls O., Antignac C., Schaefer F., Salomon R.: Prevalence of mutations in renal development genes in children with renal hypoplasia: results of the ESCAPE Study. J. Am. Soc. Nephrol., 2006; 17: 2864-2870
[PubMed] [Full Text HTML] [Full Text PDF]
[58] Weber S., Taylor J.C., Winyard K., Baker K.F., Sullivan-Brown J., Schild R., Knüppel T., Zurowska A.M., Caldas-Alfonso A., Litwin M., Emre S., Ghiggeri G.M., Bakkaloglu A., Mehls O., Antignac C., Network E., Schaefer F., Burdine R.D.: SIX2 and BMP4 mutations associated with anomalous kidney development. J Am. Soc. Nephrol., 2008; 19: 891-903
[PubMed] [Full Text HTML] [Full Text PDF]
[59] Woolf A.S.: A molecular and genetic view of human renal and urinary tract malformations. Kidney Int., 2000; 58: 500-512
[PubMed] [Full Text HTML] [Full Text PDF]
[60] Wu X.R., Kong X.P., Pellicer A., Kreibich G., Sun T.T.: Uroplakins in urothelial biology, function, and disease. Kidney Int., 2009; 75: 1153-1165
[PubMed]
[61] Yosypiv I.V.: Renin-angiotensin system in ureteric bud branching morphogenesis; insights into mechanisms. Pediatr. Nephrol., 2011; 26: 1499-1512
[PubMed]
[62] Yu J.: Wnt signaling and renal medulla formation. Pediatr. Nephrol., 2011; 26: 1553-1557
[PubMed]
[63] Żurowska A., Zagożdżon I., Bałasz I., Boguszewska A., Prokurat S., Pietrzyk J.A., Drożdż D., Szczepańska M., Stefaniak E, Jander A., Roszkowska-Blaim M., Ziółkowska H., Makulska I., Kołłątaj B., Jarmoliński T., Siteń G., Stankiewicz R., Wierciński R.: Wady dróg moczowych jako przyczyna przewlekłej choroby nerek u dzieci według Polskiego Rejestru Dzieci Leczonych Nerkozastępczo w latach 2000-2007. Pol. Merk. Lek., 2008; 24, supl. 3: 29-31
[Abstract]
Autorki deklarują brak potencjalnych konfliktów interesów.