
Cardiotoxic consequences of ionizing radiation and anthracyclines
Waldemar M Przybyszewski 1 , Maria Wideł , Joanna Rzeszowska-Wolny
Streszczenie
Mechanizmy ubocznej toksyczności zarówno promieniowania jonizującego, jak i leków antracyklinowych, oparte są na wielu reakcjach wolnorodnikowych. Molekułami docelowymi wolnych rodników są, oprócz DNA i białek, fosfolipidy obficie występujące w strukturach błoniastych komórki. Peroksydacja lipidów w błonach kardiomiocytów może w konsekwencji prowadzić do uszkodzenia struktury i funkcji tych komórek. Dynamika uszkodzeń oksydacyjnych jest konsekwencją zarówno stopnia natężenia stanu stresu oksydacyjnego, jak i obniżonego potencjału antyoksydacyjnego. Ponieważ mięsień sercowy wykazuje niewielką obronę antyoksydacyjną, jest on szczególnie podatny na utleniające właściwości wolnych rodników generowanych zarówno przez promieniowanie, jak i cytostatyki o radiomimetycznym mechanizmie działania. Zarówno popromienna, jak i polekowa toksyczność stanowią poważny problem kliniczny, często współistniejący z obniżoną fizjologicznie lub patologicznie obroną antyoksydacyjną włączonych w zabiegi terapeutyczne tkanek prawidłowych. Obserwowane zarówno addytywne, jak i nawet synergistyczne następstwa zazwyczaj wielolekowej chemioterapii przed, w trakcie lub po napromienieniu często maskują popromienne uszkodzenia mięśnia sercowego.
Słowa kluczowe:promieniowanie jonizujące • antracykliny • kardiotoksyczność
Summary
The mechanisms of the toxic side effects of radiation and anthracyclines are generally based on free radical reactions. Target molecules for free radicals are not only DNA and proteins, but also membrane structures abundant in phospholipids. Cardiomyocyte membrane lipid peroxidation may consequently lead to structural and functional damage. The dynamics of oxidative injury result from the oxidative stress status on the one hand and lowered antioxidant defense on the other. Heart muscle is especially vulnerable to the oxidative activity of free radicals generated by both radiation and cytostatics because of its low antioxidant defense. The toxic effects of radiation and cytostatics are a serious clinical problem connected with the physiologically or pathologically reduced antioxidant defense of normal tissues involved in the therapy. The additive, even synergistic, effects of the polychemotherapy usually used before, during, and after radiation often disguise postradiation effects on cardiac muscle.
Key words:ionizing radiation • anthracyclines • cardiotoxicity
Promieniowanie jonizujące oraz niektóre cytostatyki, m.in. pochodne antracyklin, oprócz działania przeciwnowotworowego charakteryzuje kardiotoksyczność jako jedno z działań niepożądanych [2,9,65]. Ponieważ coraz powszechniej stosuje się leczenie promieniami w połączeniu z chemioterapią, należy oczekiwać zwiększonego ryzyka skumulowanego działania kardiotoksycznego obydwu czynników [2,18]. Istnieje przekonanie, że niektóre uszkodzenia mięśnia sercowego mają charakter subkliniczny i pozostają nierozpoznane lub mogą ujawniać się późno w wyniku kumulacji z działaniem innych czynników niezwiązanych z leczeniem przeciwnowotworowym [65].
Mechanizmy ubocznej toksyczności tak promieniowania jonizującego, jak i leków antracyklinowych oparte są na reakcjach wolnorodnikowych inicjujących procesy peroksydacyjne makromolekuł komórkowych [3,38]. Peroksydacja lipidów w błonach kardiomiocytów może w konsekwencji prowadzić do uszkodzenia struktury i funkcji tych komórek [34]. Informacja o peroksydacyjnym uszkodzeniu mięśnia sercowego pozwoliłaby na kontrolowanie stopnia toksyczności leczenia przeciwnowotworowego, stosowanie środków kardioochronnych oraz modyfikację schematów leczenia w celu obniżenia późnych poważnych następstw kardiotoksycznych.
Należy jednak podkreślić, że peroksydacyjne zmiany w kardiomiocytach mogą być tylko jednym z wielu patogennych czynników popromiennych i/lub polekowych powodujących dysfunkcję mięśnia sercowego.
KARDIOTOKSYCZNOŚĆ CYTOSTATYKÓW ANTRACYKLINOWYCH
Liczną grupę antybiotyków antracyklinowych należącą do często stosowanych cytostatyków w leczeniu chorób nowotworowych stanowią: daunorubicyna (DNR), doksorubicyna (DOX), epirubicyna (EPI), idarubicyna (IDA) (ryc. 1). Należy zaznaczyć, że z bardzo wielu zsyntetyzowanych związków analogowych tylko kilka zostało zaakceptowanych do stosowania w klinice [57]. Taką akceptację uzyskała epirubicyna jako alternatywny lek wobec doksorubicyny oraz idarubicyna versus daunorubicyna.
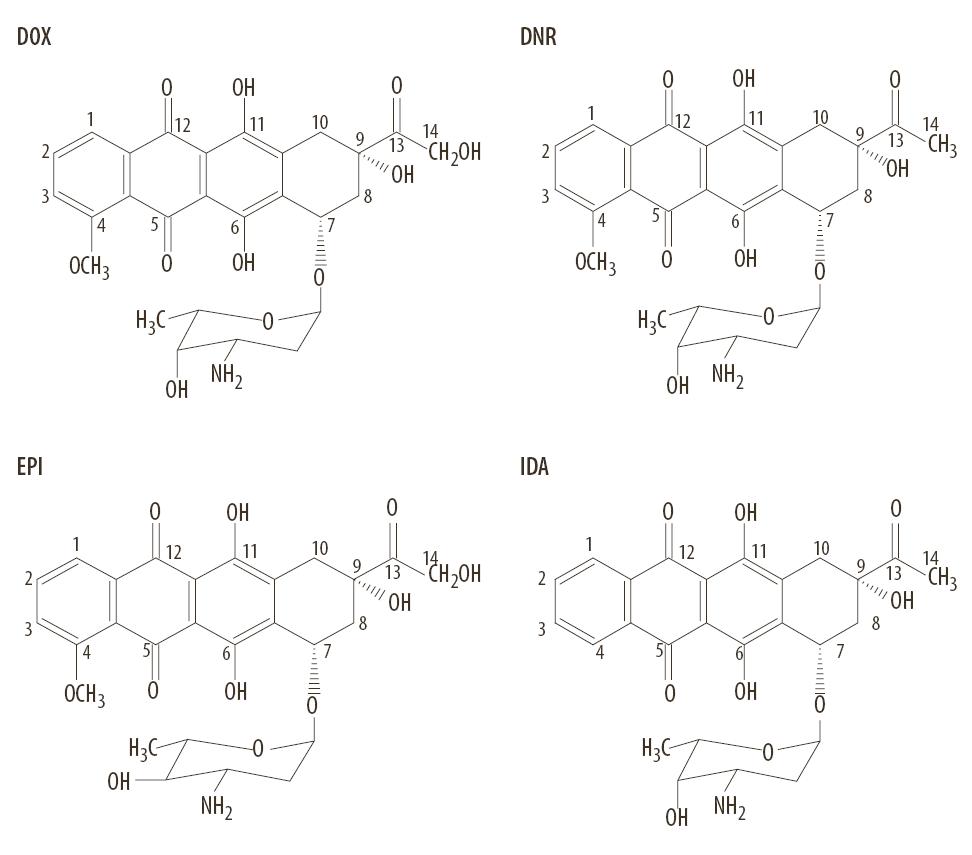
Ryc. 1. Wzory strukturalne antybiotyków antracyklinowych: DOX – doksorubicyna, DNR – daunorubicyna, EPI – epidoksorubicyna, IDA – idarubicyna
W leczeniu nowotworów znalazły również zastosowanie pirarubicyna, aklarubicyna oraz mitoksantron. Podstawowym mechanizmem działania terapeutycznego tych fazoswoistych antybiotyków jest zahamowanie aktywności enzymatycznej topoizomerazy II – DNA, syntezy RNA, oraz interkalowanie w DNA [38,57]. Wielkość dawek tych leków jest ograniczona z powodu ich kardiotoksyczności [41,57], a także mielotoksyczności [30] oraz nefrotoksyczności [20]. Wiele cytostatyków stosowanych powszechnie w chemioterapii nowotworów, takich jak: cyklofosfamid, 5-fluorouracyl, arabinozyd cytozyny, bleomycyna, taksol czy interferon wykazuje również działanie kardiotoksyczne [65]. Największe jednak zagrożenie kardiomiopatią stwarzają cytostatyki antracyklinowe [61]. Kardiomiopatia poantracyklinowa charakteryzuje się dwoma typami odziaływań, wczesnym oraz późnym. Kardiotoksyczność wczesna ujawnia się po upływie godzin lub dni od podania leku i objawia się tachykardią, nieprawidłowym obrazem EKG sygnalizującym zaburzenie funkcji lewej komory serca. W warunkach klinicznych wczesna kardiotoksyczność może wywołać późną niewydolność krążenia. Przedłużanie terapii z udziałem antracyklin może prowadzić do nieodwracalnego uszkodzenia mięśnia sercowego, którego wynikiem jest zastoinowa niewydolność krążeniowa z towarzyszącymi jej wysiękami opłucnowym i trzewnym oraz hepatomegalią i kardiomegalią [2]. Badania kliniczne wykazały, że stosowanie wielokrotnych dawek doksorubicyny przekraczające 550 mg/m2 powoduje wzrost częstotliwości występowania kardiomiopatii o 30%. Natomiast po dawkach sumarycznych nieprzekraczających 500 mg/m2 kardiomiopatię obserwuje się tylko u około 1% chorych [9]. Wiek chorych, poniżej 15 i powyżej 65 roku życia również wydaje się istotnym czynnikiem ryzyka częstości pojawiania się niewydolności krążeniowej prawokomorowej (congestive heart failure – CHF) indukowanej leczeniem doksorubicyną nawet dawką sumaryczną nieprzekraczającą 400 mg/m2 [56].
W kardiotoksycznym działaniu antracyklin uczestniczy wiele mechanizmów, takich jak: hamowanie syntezy kwasów nukleinowych oraz białek, obniżenie poziomu GSH i ATP, upośledzenie absorpcji adeniny, aminokwasów i glukozy, hamowanie aktywności enzymatycznej Na+ i K+ ATPazy, wzrost wewnątrzkomórkowego stężenia jonów wapnia, uszkodzenie mitochondriów i zaburzenie procesów oksydatywnej fosforylacji, hamowanie aktywności topoizomerazy II, wzrost stężenia histaminy i katecholamin, generowanie wolnych rodników. Wskazuje się również na upośledzenie metabolizmu sfingolipidów oraz modulację ekspresji genów [57,65,67].
Terapia nowotworów antracyklinami indukuje ogólnoustrojowy stres oksydacyjny, który zakłóca efektywność leczenia. Mediatorami stresu oksydacyjnego są aktywne formy tlenu, rodniki lipidowe, końcowe produkty peroksydacji lipidów [15] oraz aktywne formy azotu [5,60].
Mediatory stresu oksydacyjnego indukowanego przez doksorubicynę charakteryzują się m.in. działaniem proapoptotycznym. Zaobserwowano, że doksorubicyna indukuje w kardiomiocytach, in vitro, procesy apoptotyczne [4,27,28,64]. Pozostaje sprawą dyskusyjną udział tych procesów w kardiotoksycznym mechanizmie działania doksorubicyny in vivo. Wykazano, że wielokrotne iniekcje doksorubicyny powodowały krótkotrwały wzrost apoptozy komórek serca zwierząt, który obniżał się jednak przed zakończeniem terapii i pojawieniem się późnej kardiomiopatii, jakkolwiek wczesna kardiotoksyczność doksorubicyny była połączona ze wzrostem odsetka komórek ulegajacych apoptozie [6]. Jednokrotnie podana in vivo duża dawka doksorubicyny indukowała w kardiomiocytach wzrost stężenia cytochromu c oraz wzrost aktywności kaspazy 3. Wzrostowi temu towarzyszyła oligonukleosomalna i mononukleosomalna fragmentacja DNA [12]. Wzrost odsetka kardiomiocytów oraz komórek endotelialnych naczyń wieńcowych mięśnia serca ulegających apoptozie obserwowano in vivo w zależności od dawki adriamycyny [62]. Badania na innym modelu zwierzęcym wykazały natomiast, że wielokrotnie podawane dawki doksorubicyny nie miały wpływu na apoptozę kardiomiocytów [66].
Badania kardiomiocytów osób z samoistną kardiomiopatią rozstrzeniową (dilated cardiomyopathy), czy niedokrwienną chorobą serca (ischaemic heart disease) wskazywały na apoptozę jako charakterystyczną cechę ostatniej fazy niewydolności mięśnia sercowego [51].
POPROMIENNE SKUTKI USZKODZENIA SERCA
Popromienne uszkodzenie mięśnia sercowego może sprzyjać powstawaniu lub nasilać już istniejące schorzenia mięśnia sercowego. Wysiękowe zapalenie mięśnia sercowego jest częstym odczynem popromiennym (25–60%). Jest ono wynikiem uszkodzenia systemu włosowatych naczyń krwionośnych oraz limfatycznych. Najczęściej (w 80%) wysięk osierdziowy pojawia się w okresie do 2 lat po radioterapii, a następnie ustępuje. Uszkodzone napromieniowaniem miokardium charakteryzuje się rozsianym, śródmiąższowym zwłóknieniem co upośledza rozkurczową czynność serca. Pojawiająca się tkanka bliznowata w połączeniu z zanikiem kardiomiocytów powoduje ograniczenie prawidłowego rozwoju serca, zwłaszcza u osób, które w wieku dziecięcym zostały napromieniowane z przyczyn terapeutycznych [2]. W niektórych przypadkach uszkodzenie serca ma charakter nieodwracalnego i postępującego zwłóknienia [9,22,65]. U chorych leczonych napromieniowaniem śródpiersia miażdzycowe stwardnienie naczyń wieńcowych serca jest późną odpowiedzią na terapię promieniami. Uważa się, że jednym z zasadniczych czynników patogenezy popromiennego uszkodzenia serca może być uszkodzenie struktury i funkcji sieci naczyń włosowatych. Niedokrwienie będące konsekwencją zaniku mikrokrążenia prowadzi do atrofii i zwłóknienia powodując kardiomiopatię [2,8].
Analiza dynamiki transkrypcji genów transformującego czynnika wzrostu beta-1 (TGF-beta 1) i prokolagenów typu I i III w kardiomiocytach lewej komory po napromieniowaniu in vivo serca szczurów dawkami 15 Gy, 20 Gy i 25 Gy, wykazała wczesny, do 12 godzin, szybki dwufazowy wzrost poziomu mRNA TGF-beta 1. Poziom ten normalizował się po upływie jednego miesiąca i nie miał wpływu na ekspresję mRNA obu typów prokolagenów, która powoli, systematycznie wzrastała do 16 miesiecy po napromienieniu [31]. Napromieniowanie serca pojedynczymi dawkami 15 Gy i 20 Gy powodowało też wzrost akumulowania się czynnika von Willebranda sygnalizując uszkodzenie endotelium poprzedzające rozwój zwłóknień w mięśniu sercowym [10].
Popromienne zakłócenie motoryki serca powoduje ujawnienie się mechanizmów kompensujących pracę serca. Odpowiedzią na to zakłócenie jest pobudzenie sympatycznego układu nerwowego, zmienny poziom katecholamin oraz zmiana w gęstości receptorów adrenergicznych [9]. Można z dużym prawdopodobieństwem przyjąć, że zmiany te odzwierciedlają zwiększoną stymulację pracy serca przez układ nerwowy celem przeciwdziałania upośledzeniu czynności serca.
Popromienne uszkodzenie serca może mieć charakter progresywny, przy czym zależność między nasileniem tego uszkodzenia i dawką całkowitą oraz dawką frakcyjną nie jest dostatecznie precyzyjnie określona [52,53].
Sugerowane są, ogólnie rzecz ujmując, dwa mechanizmy uszkodzenia tkanki przez promieniowanie jonizujące [55]. Pierwszym jest bezpośrednia jonizacja składników komórki. Drugi mechanizm – pośredni, stanowi radioliza wewnątrzi zewnątrzkomórkowej wody. Uważa się, że popromienna modyfikacja organizacji systemu błon komórkowych ma znaczący udział w tworzeniu radiobiologicznych efektów promieniowania jonizującego [3,54] (ryc.2).
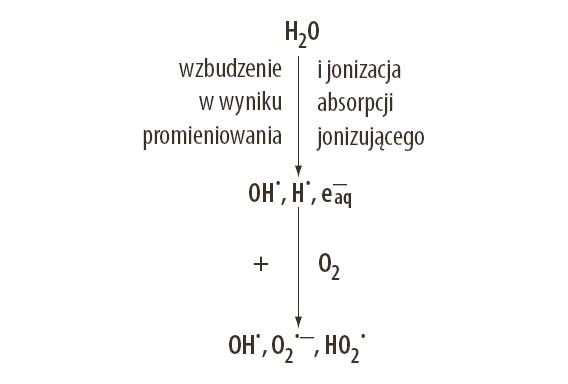
Ryc. 2. Ogólny schemat powstawania wolnych rodników w wyniku radiolizy wody
Popromienne procesy peroksydacji lipidów są wynikiem zarówno oddziaływania bezpośredniego promieniowania na fazę lipidową, jak również pośredniego działania i pojawienia się wolnych rodników. Ponieważ wykazano, że obecność wody znamiennie decyduje o wzroście stopnia peroksydacji lipidów, to sugeruje się, że radioliza wody może stanowić podstawowy mechanizm inicjujący procesy peroksydacyjne w błonach [55].
Interesująca wydaje się również sugerowana możliwość popromiennej aktywacji enzymatycznego systemu oksydoreduktazy ksantynowej (XOR), generujacego wolne rodniki w radiolitycznie uszkodzonej tkance, co przyczyniałoby się do zwielokrotnienia oraz utrwalenia późnych efektów popromiennych [26].
KARDIOTOKSYCZNE NASTĘPSTWA KOJARZONEGO STOSOWANIA PROMIENIOWANIA I ANTRACYKLIN
Wyniki badań morfologiczno-histologicznych na zwierzętach wykazały, że efekt kojarzenia obu czynników kardiotoksycznych jest nie tylko addytywny, ale prawdopodobnie synergistyczny. Badania kliniczne dostarczyły podobnych wniosków. Jest również możliwe, że efekt taki może być wynikiem „dawki zapamiętanej” (recall phenomenon). Wiąże się to z procesem powtórnej aktywacji efektu popromiennego przez cytostatyki antracyklinowe w obszarze uprzednio napromienionej tkanki [7]. Wyniki badań doświadczalnych na myszach wskazują, że zarówno adriamycyna jak i w mniejszym stopniu aktynomycyna D podane zwierzętom po kilku tygodniach lub miesiącach po napromieniowaniu klatki piersiowej powodują wzmożenie efektu popromiennego, ujawniając tym subkliniczne procesy zachodzące w okresie latencji zmian popromiennych [59]. Mechanizmy tego procesu nie są jednak w pełni zbadane i wyjaśnione.
Wyniki badań efektów kardiotoksycznych łączonej terapii promieniami i doksorubicyną na modelu zwierzęcym nie wykazały jednak działania synergistycznego w odniesieniu do zmian funkcjonalnych, uszkodzenia peroksydacyjnego serca czy zaburzenia endogennej ochrony antyoksydacyjnej [18]. Badania własne wykazały, że kojarzone napromieniowania serca z dootrzewnową iniekcją farmorubicyny nie wywoływało zwiększonej peroksydacji w tkance serca w odniesieniu do grupy zwierząt otrzymujących tylko farmorubicynę w analogicznych dawkach i sekwencji podawania. Natomiast działanie peroksydacyjne było wzmożone w porównaniu do grupy zwierząt tylko napromieniowanych [46]. Kojarzenie napromieniowania i farmorubicyny również nie powodowało wzrostu aktywności kinazy kreatynowej (CK) w stosunku do aktywności mierzonej po zadziałaniu każdego z tych czynników samodzielnie. Relatywnie mniejszy wzrost aktywności izoenzymu CK-MB po skojarzonym działaniu obu rodnikorodnych czynników był prawdopodobnie wynikiem rekombinacji wolnych rodników i obniżeniem dynamiki procesów peroksydacyjnych [45]. Jednak wzrost aktywności CK-MB, izoenzymu swoistego dla mięśnia sercowego, w grupach napromieniowanych czy traktowanych farmorubicyną (niezależnie od schematu podawania) był wielokrotnie większy niż wzrost aktywności całkowitej kinazy kreatynowej w odniesieniu do odpowiednich grup kontrolnych. Obserwacje zwierząt powyżej sześciu miesięcy ujawniły toksyczność objawiającą się jako stagnacja lub utrata masy ciała szczurów. Przypadki zgonów zwierząt były również obserwowane, a pośmiertne badania sekcyjne ujawniały powiększone serce i wątrobę oraz wysięki w jamie otrzewnowej i opłucnowej. Działania te występowały częściej w grupach zwierząt, gdzie napromieniowanie serca łączono z podawaniem farmorubicyny.
Uszkodzenie serca wywołane doksorubicyną różni się zdecydowanie od uszkodzenia popromiennego, ale uzupełnia się z punktu widzenia działań patofizjologicznych i są one zależne od wielkości dawki. Doksorubicyna uszkadza bezpośrednio kardiomiocyty, natomiast promieniowanie uszkadza system naczyniowy, co prowadzi do śródmiąższowego zwłóknienia [9].
Jednak najświeższe doniesienia dowodzą, że antracykliny powodują także uszkodzenie śródbłonka naczyń krwionośnych, co wykazano w badaniach in vitro [39,40,62] oraz in vivo [62]. Kliniczne badania pilotowe na niewielkiej grupie chorych nowotworowo (średnia wieku 15,5 lat) leczonych doksorubicyną w dawce nie mniejszej niż 300 mg/m2 oraz równoważną jej dawką daunorubicyny 350 mg/m2 wykazały uszkodzenie naczyń krwionośnych przejawiające się obniżeniem aktywności wazomotorycznej utrzymującej się do roku [13]. Uważa się, że dysfunkcja endotelium może stanowić wczesny etap rozwoju popromiennej i/lub polekowej miażdżycy naczyń wieńcowych. Interesujące wydaje się również zbadanie wpływu innych cytostatyków, stosowanych w skojarzeniu z antracyklinami w leczeniu nowotworów, na aktywność wazomotoryczną naczyń krwionośnych serca.
KARDIOTOKSYCZNOŚĆ PEROKSYDACYJNA
Kardiotoksyczności doksorubicyny przypisuje się zarówno jej zdolność do generowania aktywnych form tlenu (ROS), jak i inicjowania uszkodzeń oksydacyjnych za pośrednictwem jonów żelaza [36,57]. Obserwowano jednak w kardiomiocytach antyoksydacyjny sposób działania antracyklin przez stymulację ekspresji ferrytyny, białka magazynującego jony żelaza, co w konsekwencji zapobiegało uszkodzeniom indukowanym przez dodane do hodowli jony żelaza [16]. Pojedyncze doniesienia sugerują też, że tlenowa degradacja antracyklin do kwasów ftalowych zapobiega peroksydacji lipidów [11].
Uważa się, że alkoholowe metabolity antracyklin biorą udział w powstaniu kardiotoksyczności późnej. Zahamowanie bowiem aktywności reduktazy karbonylowej (EC 1.1.1.184) prowadzi do kardioochrony zapobiegając enzymatycznej konwersji doksorubicyny do doksorubicinolu [42]. Wykazano, że kardiotoksyczność doksorubicyny jest połączona ze wzrostem stężenia końcowych aldehydowych produktów peroksydacji lipidów, które pojawiają się w tkance serca [1,34]. We krwi pobranej z zatoki wieńcowej serca, od pacjentów nowotworowych przed rozpoczęciem leczenia doksorubicyną stwierdzono kilkukrotnie wyższy poziom peroksydacji lipidów w porównaniu z krwią pobraną z tętnicy udowej. Infuzja doksorubicyny znosiła różnice w poziomie peroksydacji. Należy dodać, że poziom peroksydacji lipidów we krwi pacjentów nienowotworowych z dysfunkcją zastawek czy arytmią serca nie wykazywał takich różnic [37]. Jednakże we krwi tętniczej pacjentów z niewydolnością krążeniową (CHF) poziom nienasyconych aldehydów m.in. 4-hydroksynonenalu (4HNE) [34] i dialdehydu malonowego (MDA) we krwi obwodowej [19] był wyższy w porównaniu z grupą pacjentów kontrolnych. Wyniki takich badań wykazały, że zarówno niewydolność krążeniowa, jak i obecność nowotworu będące uogólnionym stanem stresu oksydacyjnego stymulują ustrojowe procesy peroksydacyjne. Zastosowanie doksorubicyny obniżało poziom peroksydacji lipidów u pacjentów nowotworowych leczonych tym lekiem [37].
Natężenie stresu oksydacyjnego, będące wypadkową tak zwiększonego powstawania wolnych rodników jak i niewielką ochroną antyoksydacyjną serca, stanowi ważny element patogenezy niewydolności mięśnia sercowego. Czynnościowe polekowe skutki zmian strukturalnych w komórce pozostają jednak nie do końca wyjaśnione. Wiele prac wskazuje na to, że uszkodzenie komórek mięśnia sercowego w następstwie reakcji wolnorodnikowych indukowane przez doksorubicynę (adriamycynę) zachodzi w wyniku peroksydacji struktur błoniastych [34,63].
Generowanie wolnych rodników przez doksorubicynę jest wynikiem działania dwóch mechanizmów. Pierwszy mechanizm stanowi reakcja redukcji doksorubicyny do wolnego rodnika semichinonowego, w której pośredniczą komórkowe enzymy oksydoredukcyjne. W wyniku reakcji rodnika semichinonowego z tlenem pojawiają się: anionorodnik ponadtlenkowy, rodnik hydroksylowy oraz nadtlenek wodoru. Drugi mechanizm generowania wolnych rodników jest konsekwencją chemicznej struktury doksorubicyny warunkującej chelatowanie jonów metali, w tym jonów żelaza. Powstały kompleks doksorubicyna-żelazo podlega chemicznej redukcji z udziałem tlenu do nadtlenku wodoru oraz rodnika wodorotlenowego [38,57]. Wykazano in vitro, że doksorubicyna stymuluje w kardiomiocytach syntezę tlenku azotu (NO), który może reagować z anionorodnikiem ponadtlenowym, co prowadzi do pojawienia się nadtlenoazotynu (peroxynitrite – ONOO–) [5]. W badaniach in vivo obserwowano procesy nitrowania białek kurczliwych, które wybiórczo modyfikowały strukturę miofibryli w kardiomiocytach powodując dysfunkcję mięśnia sercowego [60]. Należy podkreślić, że w mechanizmie kardiotoksycznym antracyklin, znaczącą rolę pełnią jony żelaza, które są katalizatorami reakcji wolnorodnikowych [36,57] (ryc. 3).
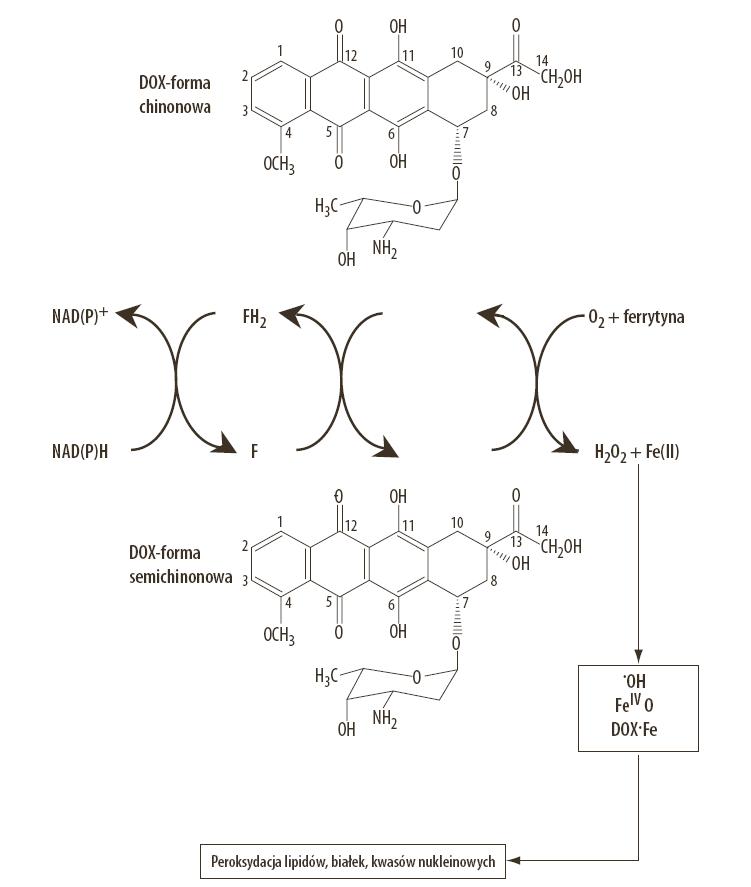
Ryc. 3. Ogólny schemat powstawania aktywnych form tlenu i wolnych rodników w wyniku enzymatycznej redukcji doksorubicyny oraz redoksowej aktywności kompleksu doksorubicyna-żelazo; F – enzymy flawoproteinowe, Dox-Fe – kompleks doksorubicyna/żelazo, FeIV= O – jon ferrylowy
Wynikiem mechanizmów rodnikowych i procesów peroksydacyjnych, tak w działaniu leków antracyklinowych [34], jak i promieniowania jonizującego [3] jest pojawienie się wielu toksycznych pośrednich i końcowych produktów peroksydacji lipidów, charakteryzujących się właściwościami mutagennymi i teratogennymi [67]. Peroksydacja lipidów systemu błon komórkowych prowadzi do pojawienia się nadtlenków lipidów i toksycznych aldehydów, w tym i dialdehydu malonowego, który w reakcji barwnej z kwasem ditiobarbiturowym tworzy kompleks barwny określany mianem TBA-RS [44]. Wodoronadtlenki lipidów oraz substancje reagujące z kwasem ditiobarbiturowym wykrywano we krwi pacjentów poddawanych działaniu agresywnej radiochemioterapii [14] czy polichemioterapii [33].
Badania własne in vivo ujawniły, że napromieniowanie serca szczura dawkami promieniowania gamma 20 Gy [47] lub 4×5 Gy [46] po kilku dniach powodowało znamienny wzrost stężenia produktów peroksydacji lipidów nie tylko w sercu, ale i w surowicy. Pojawienie się TBA-RS w surowicy jest prawdopodobnie wynikiem wypłukiwania z tkanki serca oraz napromieniowania krwi przepływającej przez serce, w trakcie ekspozycji na wiązkę promieniowania. Wielokrotnie podane dawki promieniowania (4×5 Gy) indukowały większe stężenia TBA-RS zarówno w sercu, jak i surowicy w porównaniu do dawki jednorazowej 20 Gy. Efekt ten jest prawdopodobnie związany z procesami rekombinacji i inaktywacji wolnych rodników po dużych dawkach promieniowania gamma i tym samym obniżonej dynamice popromiennych procesów peroksydacji lipidów [47].
Peroksydacja lipidów w błonach komórkowych powoduje zaburzenie integralności błon, co prowadzi do wycieku enzymów [44]. Wyniki badań własnych aktywności kinazy kreatynowej w surowicy po napromieniowaniu serca szczurów in vivo zarówno dawką pojedynczą 20 Gy [47], jak i frakcjonowaną 4×5 Gy [46] wykazały wzrost aktywności enzymu w ciągu kilku pierwszych dni. Ponieważ dawki promieniowania ogniskowano na mięsień sercowy, dlatego sądzimy, że podwyższona aktywność enzymów obserwowana w surowicy była wynikiem peroksydacyjnego uszkodzenia systemu błon komórkowych kardiomiocytów. Celem określenia jaką część mierzonej aktywności kinazy kreatynowej stanowiła właściwa tkance serca aktywność, oznaczano aktywność izoenzymu CK-MB swoistego dla mięśnia sercowego. Uszkodzenie mięśnia sercowego mierzone wzrostem aktywności CK-MB było statystycznie znamienne w zakresie stosowanych dawek promieniowania [45].
Aktywność nie tylko kinazy kreatynowej (CK) czy aminotransferazy asparaginianowej (AST), lecz także dehydrogenazy mleczanowej (LDH), aminotransferazy alaninowej (ALT), dehydrogenzy alfa-hydroksymaślanu (alpha-HBDH), oznaczano w osoczu i w lewej komorze serca szczurów po napromieniowaniu pojedynczą dawką 20 Gy [21]. Obserwowano wzrost aktywności CK, LDH, AST i alpha-HBDH w osoczu w czasie pierwszej doby po napromieniowaniu. Aktywność ALT pozostawała bez zmian. Poziom aktywności tych enzymów oznaczanych systematycznie w tkance serca w czasie kilkunastu dni po napromieniowaniu nie wykazywał różnic w porównaniu z grupą kontrolną. Jednakże szybki spadek aktywności w ciągu pierwszych kilkunastu godzin po napromieniowaniu był znaczący. Podwyższonej aktywności enzymatycznej w osoczu towarzyszyła obniżona aktywność badanych enzymów w sercu [21]. Tak duża pojedyncza dawka promieniowania (20 Gy) powoduje nieodwracalne uszkodzenie serca oraz redukuje czas przeżycia zwierząt, co obserwowano we własnych eksperymentach [47].
Adriamycyna podawana królikom w dawkach wielokrotnych przez dłuższy okres powodowała wzrost stężenia wodoronadtlenków lipidów tylko w sercu, natomiast nie stwierdzano produktów peroksydacji lipidów w mózgu, wątrobie i mięśniach szkieletowych. Korelacja między intensywnością chemoluminescencji wodoronadtlenków i uszkodzeniem serca była statystyczne znamienna [32]. Doksorubicyna podana szczurom dootrzewnowo powodowała wczesny, znaczący wzrost stężenia aldehydów: heksanalu, heptanalu, oktanalu, nonanalu, propanalu, butanalu, pentanalu, furfuralu, dialdehydu malonowego, 4-hydroksynonenalu oraz produktów ich metabolizmu określanych mianem acyloin (hydroksyheptanonu, hydroksyoktanonu, hydroksynonanonu, hydroksydekanonu, hydroksydodekanonu) w osoczu i tkance serca [34]. Interesujące wydają się wyniki uzyskane z badań nad bezpośrednim upośledzającym wpływem 4-HNE na dynamikę kurczliwości kardiomiocytów wyizolowanych z mięśnia sercowego [1]. Aldehydy te, reagując z molekułami komórkowymi, zmieniają biochemiczny status komórki, co prowadzi do jej uszkodzenia.
Badania własne, in vivo, nad wczesnym peroksydacyjnym działaniem farmorubicyny, leku stosowanego klinicznie jako cytostatyk alternatywny względem doksorubicyny wykazały, że dawka 10 mg/kg m.c. podana jednorazowo [47] lub w dawce podzielonej (4×2,5 mg/kg m.c.) [46] powodowała u szczurów znamienny wzrost stopnia peroksydacji mierzony poziomem TBA-RS w sercu i w surowicy w ciągu kilku pierwszych dni. Ponadto wzrost aktywności izoenzymu sercowego kinazy kreatynowej (CK-MB) po dawkach powtarzanych farmorubicyny był wyższy w porównaniu do aktywności po dużej jednorazowej dawce leku [45].
KARDIOOCHRONA
Metody ochrony mięśnia sercowego opierają się nie tylko na molekularnych mechanizmach toksycznego działania promieniowania i leków antracyklinowych, ale również na sposobie podawania leku, doborze odpowiednich nośników, a nawet wykorzystaniu fizycznego cyklu funkcjonowania serca.
Ochronne działanie antyoksydantów potwierdza, że kardiotoksyczność związana zarówno z chemioterapią, jak i radioterapią nowotworów jest wynikiem stanu stresu oksydacyjnego wywoływanego przez te czynniki [9].
Przeciwutleniające działanie witaminy E, znanego i uznanego antyoksydanta [23,43] wykazano w badaniach własnych [46] przez statystycznie znamienne, zahamowanie powstawanie TBA-RS zarówno po napromieniowaniu serca i po podaniu farmorubicyny, jak również po skojarzonym działaniu promieniowania i farmorubicyny. Jednak ochronne działanie witaminy E nie było tak jednoznaczne jeżeli analizowano aktywność kinazy kreatynowej oznaczanej w surowicy. Zwłaszcza po napromieniowaniu serca witamina E pobudzała aktywność kinazy kreatynowej. Witamina E podawana zwierzętom otrzymującym farmorubicynę znamiennie normalizowała aktywność enzymu. Stosowanie tej witaminy u zwierząt poddanych skojarzonemu działaniu promieniowania i cytostatyku również powodowało znamienną normalizację aktywności enzymatycznej, jednak obniżenie tej aktywności poniżej wartości kontrolnych może świadczyć o zwiększonym uszkodzeniu kardiomiocytów, mimo obecności witaminy E.
Kardioprotekcyjnymi właściwościami charakteryzuje się Neoton (fosforan kreatyny). Jest to związek fizjologiczny znajdujący się w metabolicznie aktywnych tkankach, takich jak mięsień sercowy. Ważną cechą tej substancji jest przeciwdziałanie peroksydacji oraz stabilizowanie błon komórkowych. Wyniki doświadczeń in vitro, w których stosowano Neoton w roztworach kardioplegicznych wykazały, że zapobiegał on zmianom czynnościowym serca perfundowanego nadtlenkiem wodoru. Hamował powstawanie dialdehydu malonowego w sercu, czego nie powodowały rozdzielnie stosowane kreatyna oraz fosforan nieorganiczny. Zapobiegał także wyciekowi enzymów z kardiomiocytów, zabezpieczając integralność systemu błonowego komórki [50].
Wyniki badań własnych w niewielkim stopniu potwierdziły in vivo antyoksydacyjne działanie Neotonu wobec peroksydacyjnych działań małych, wielokrotnie podanych dawek promieniowania oraz farmorubicyny [48].
Jednym z efektów działania zarówno promieniowania gamma [49], jak i adriamycyny [36] jest proces uwalniania jonów żelaza zmagazynowanego w komórkowych złożach ferrytyny. Wolne jony żelaza biorąc udział w reakcjach oksydacyjnych stymulują powstawanie dodatkowej puli wolnych rodników, co dynamizuje procesy peroksydacji lipidów.
Zastosowanie leków, których mechanizm działania opiera się na wychwytywaniu dwuwartościwych jonów metali, powodowałoby obniżenie dynamiki formowania się wolnych rodników. Kardioksan okazał się w badaniach in vitro i in vivo lekiem obniżającym kardiotoksyczność antybiotyków antracyklinowych [17]. Również niektóre kontrolowane doświadczenia kliniczne potwierdziły kardioochronne działania kardioksanu. Jednakże rutynowe stosowanie tego leku w klinice wymaga dalszych badań [41]. Należy dodać, że podawany sam powodował mielosupresję oraz wzrost częstości pojawiania się mikrojąder w erytrocytach polichromatycznych [29]. Powodował również pogłębienie cytotoksycznego działania promieniowania in vitro, jak i in vivo [47].
Wyniki badań własnych in vivo wykazały, że kardioksan podany sam powoduje statystycznie znamienny wzrost stężenia produktów peroksydacji lipidów (TBA-RS) zarówno w sercu jak i surowicy. Zastosowanie kardioksanu w połączeniu z jednorazowymi dawkami promieniowania lub farmorubicyny w niewielkim stopniu wpływało na obniżenie poziomu TBA-RS. Efekty normalizującego działania kardioksanu na aktywność kinazy kreatynowej nie były również obserwowane. Przeciwnie, kardioksan wzmagał uszkodzenie serca powodowane przez promieniowanie lub farmorubicynę mierzone aktywnością enzymu [47]. Wyniki badań in vitro wykazały, że produkt hydrolizy kardioksanu stymuluje powstawanie rodników hydroksylowych [25,58]. Proces ten prawdopodobnie stanowił w przeprowadzonych badaniach własnych in vivo dodatkowe źródło agresywnych rodników dynamizujących peroksydacyjne działanie promieniowania oraz farmorubicyny.
Interesującym sposobem na ograniczenie uszkodzenia mięśnia sercowego w trakcie radioterapii wydaje się technika synchronizacji wiązki promieniowania z fazami cyklu serca, tj.pulsacyjnego napromieniania w chwili, gdy serce znajduje się w stanie względnej hipoksji [24].
Powolna, wielogodzinna, ciągła infuzja doksorubicyny powodowała mniejszą kardiotoksyczność w porównaniu z podawaniem standardowym nie wpływając na obniżenie odpowiedzi terapeutycznej guza. Jednakże taka technika podawania, jak wykazały badania, powodowała wzrost uszkodzeń DNA w leukocytach krwi obwodowej. Innym sposobem niwelowania kardiotoksyczności jest technika stosowania antracyklin w postaci liposomów, immunoliposomów, opłaszczonych polietylenoglikolem w postaci kopolimeru albo też pochodnych morfolinylowych lub disacharydowych. Innym, ważnym zagadnieniem pozostaje synteza chemiczna kolejnych mało kardiotoksycznych antracyklin [38,57].
PIŚMIENNICTWO
[1] Aberle N.S.2nd., Picklo M.J.Sr., Amarnath V., Ren J.: Inhibition of cardiac myocyte contraction by 4-hydroxy-trans-2-nonenal. Cardiovasc. Toxicol., 2004; 4: 21-28
[PubMed]
[2] Adams M.J., Lipshultz S.E.: Pathophysiology of anthracycline- and radiation-associated cardiomyopathies: Implications for screening and prevention. Pediatr. Blood Cancer., 2005; 44: 600-606
[PubMed]
[3] Agrawal A., Kale R.K.: Radiation induced peroxidative damage: mechanism and significance. Indian J. Exp. Biol., 2001; 39: 291-309
[PubMed]
[4] Akao M., O’Rourke B., Teshima Y., Seharaseyon J., Marban E.: Mechanistically distinct steps in the mitochondrial death pathway triggered by oxidative stress in cardiac myocytes. Circ. Res., 2003; 92: 186-194
[PubMed] [Full Text HTML] [Full Text PDF]
[5] Aldieri E., Bergandi L., Riganti C., Costamagna C., Bosia A., Ghigo D.: Doxorubicin induces an increase of nitric oxide synthesis in rat cardiac cells that is inhibited by iron supplementation. Toxicol. Appl. Pharmacol., 2002; 185: 85-90
[PubMed]
[6] Arola O.J., Saraste A., Pulkki K., Kallajoki M., Parvinen M., Voipio-Pulkki L-M.: Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res., 2000; 60: 1789-1792
[PubMed] [Full Text HTML] [Full Text PDF]
[7] Azria D., Magne N., Zouhair A., Castadot., Culine S., Ychou M., Stupp R., Van Houtte P., Dubois J-B., Ozsahin M.: Radiation recall: Awell recognized but neglected phenomenon. Cancer Treat. Rev., 2005; 31: 555-570
[PubMed]
[8] Basavaraju S.R., Easterly C.E.: Pathophysiological effects of radiation on atherosclerosis development and progression, and incidence of cardiovascular complications. Med. Phys., 2002; 29: 2391-2403
[PubMed]
[9] Berry G.J., Jorden M.: Pathology of radiation and anthracyclines cardiotoxicity. Pediatr. Blood Cancer, 2005; 44: 630-637
[PubMed]
[10] Boerma M., Kruse J.J., van Loenen M., Klein H.R., Bart C.I., Zurcher C., Wondergem J.: Increased deposition of von Willebrand factor in the rat heart after local ionizing irradiation. Strahlenther. Onkol., 2004; 180: 109-116
[PubMed]
[11] Cartoni A., Menna P., Salvatorelli E., Braghiroli D., Giampietro R., Animati F., Urbani A., Del Boccio P., Minotti G.: Oxidative degradation of cardiotoxic anticancer anthracyclines to phtalic acids: novel function for ferrylmyoglobin?. J. Biol. Chem., 2004; 279: 5088-5099
[PubMed] [Full Text HTML] [Full Text PDF]
[12] Childs A.C., Phaneuf S.L., Dirks A.J., Phillips T., Leeuwenburgh C.: Doxorubicin treatment in vivo causes cytochrome c release and cardiomycyte apoptosis as well as increased mitochondrial efficiency, superoxide dismutase activity and Bcl-2: Bax ratio. Cancer Res., 2002; 62: 4592-4598
[PubMed] [Full Text HTML] [Full Text PDF]
[13] Chow A.Y., Chin C., Dahl G., Rosenthal D.N.: Anthracyclines cause endothelial injury in pediatric cancer patients: A pilot study. J. Clin. Oncol., 2006; 24: 925-928
[PubMed]
[14] Clemens M.R., Ladner C., Schmidt H., Ehninger G., Einsele H., Buhler E., Waller H.D., Gey K.F.: Decreased essential antioxidants and increased lipid hydroperoxides following high-dose radiochemotherapy. Free Radic. Res. Commun., 1989; 7: 227-232
[PubMed]
[15] Conklin K.A.: Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther., 2004; 3: 294-300
[PubMed] [Full Text PDF]
[16] Corna G., Santambrogio P., Minotti G., Cairo G.: Doxorubicin paradoxically protects cardiomiocytes against iron-mediated toxicity. J. Biol. Chem., 2004; 279: 13738-13745
[PubMed] [Full Text HTML] [Full Text PDF]
[17] Cvetkovic R.S., Scott L.J.: Dexrazoxane: a review of its use for cardioprotection during anthracycline chemotherapy. Drugs, 2005; 65: 1005-1024
[PubMed]
[18] Dalloz F., Maingnon P., Cottin Y., Briot F., Horiot J.C., Rochette L.: Effects of combined irradiation and doxorubicin treatment on cardiac function and antioxidant defenses in the rat. Free Radic. Biol. Med., 1999; 26: 785- 800
[PubMed]
[19] Diaz-Velez C.R., Garcia-Castineiras S., Mendoza-Ramos E., Hernandez-Lopez E.: Increased malondialdehyde in peripheral blood of patients with congestive heart failure. Am. Heart J., 1996; 131: 146-152
[PubMed]
[20] Dzięgiel P., Suder E., Surowiak P., Jethon Z., Rabczyński J., Januszewska L., Sopel M., Zabel M.: Role of exogenous melatonin in reducing the nephrotoxic effect of daunorubicin and doxorubicin in the rat. J. Pineal Res., 2002; 33: 95-100
[PubMed]
[21] Franken N.A., Strootman E., Hollaar L., van der Laarse A., Wondergem J.: Myocardial enzyme activities in plasma after whole-heart irradiation in rats. J. Cancer Res. Clin. Oncol., 2000; 126: 27-32
[PubMed]
[22] Gaya A.M., Ashford R.F.: Cardiac complications of radiation therapy. Clin. Oncol., (R. Coll. Radiol.), 2005; 17: 153-159
[PubMed]
[23] Gaziano J.M.: Vitamin E and cardiovacular disease: observational studies. Ann. N. Y. Acad. Sci., 2004; 1031: 280-291
[PubMed]
[24] Gladstone D.J., Flanagan M.F., Southworth J.B., Hadley V., Thibualt M.W., Hug E.B., Hoopes P.J.: Radiation-induced cardiomyopathy as a function of radiation beam gating to the cardiac cycle. Phys. Med. Biol., 2004; 49: 1475-1484
[PubMed]
[25] Hasinoff B.B.: NADPH-cytochrome-P450 reductase promotes hydroxyl radical production by the iron complex of ADR-925, the hydrolysis product of ICRF-187 (dexrazoxane). Free Radical. Res., 1995; 22: 319-325
[PubMed]
[26] Kale R.K.: Post-irradiation free radical generation: evidence from the conversion of xanthine dehydrogenase into xanthine oxidase. Indian J. Exp. Biol., 2003; 41: 105-111
[PubMed]
[27] Kang Y.J., Zhou Z.X., Wang G.W., Buridi A., Klein J.B.: Suppression by metallothioein of doxorubicin-induced cardiomyocyte apoptosis through inhibition of p38 mitogen-activated protein kinases. J. Biol. Chem., 2000; 275: 13690-13698
[PubMed] [Full Text HTML] [Full Text PDF]
[28] Kim Y., Ma A.G., Kitta K., Fitch S.N., Ikeda T., Ihara Y., Simon A.R., Evans T., Suzuki Y.J.: Anthracycline-induced suppression of GATA-4 transcription factor: implication in the regulation of cardiac myocyte apoptosis. Mol. Pharmacol., 2003; 63: 368-377
[PubMed] [Full Text HTML] [Full Text PDF]
[29] Koning J., Palmer P., Franks C.R., Mulder D.E., Speyer J.L., Green M.D, Hellmann K.: Cardioxane ICRF-187 towards anticancer drug specificity through selective toxicity reduction. Cancer Treat. Rev., 1991; 18: 1-19
[PubMed]
[30] Kroschinsky F., Schleyer E., Renner U., Schimming C., Schimmelpfennig C., Bornhausen M., Illmer T., Trumper L., Ehninger G., Schaich M.: Increased myelotoxicity of idarubicin: is there a pharmacological basis? Results of a pharmacokinetic and an in vitro cytotoxicity study. Cancer Chemother.Pharmacol., 2004; 53: 61-67
[PubMed]
[31] Kruse J.J., Bart C.I., Visser A., Wondergem J.: Changes in transforming growth factor-beta (TGF-beta 1), procollagen types I and II mRNA in the rat heart irradiation. Int. J. Radiat. Biol., 1999; 75: 1429-1436
[PubMed]
[32] Llesuy S.F., Milei J., Gonzalez Flecha B.S., Boveris A..: Myocardial damage induced by doxorubicins hydroperoxide-initiated chemiluminescence and morphology. Free Rad. Biol. Med., 1990; 8: 259-264
[PubMed]
[33] Look M.P., Musch E.: Lipid peroxides in the polychemotherapy of cancer patients. Chemotherapy, 1994; 40: 8-15
[PubMed]
[34] Luo X., Evrovsky Y., Cole D., Trines J., Benson L.N., Lehotay D.C.: Doxorubicin-induced acute changes in cytotoxic aldehydes, antioxidant status and cardiac function in the rat. Biochim. Biophys. Acta, 1997; 1360: 45-52
[PubMed]
[35] Mak S., Lehotay D.C., Yazdanpanah M., Azevedo E.R., Liu P.P., Newton G.E.: Unsaturated aldehydes including 4-OH-nonenal are elevated in patients with congestive heart failure. J. Card. Fail., 2000; 6: 108-114
[PubMed]
[36] Minotti G., Cairo G., Monti E.: Role of iron in anthracycline cardiotoxicity: new tunes for an old song? FASEB J., 1999; 13: 199-212
[PubMed] [Full Text HTML] [Full Text PDF]
[37] Minotti G., Mancuso C., Frustaci A., Mordente A., Santini S.A., Calafiore A.M., Liberi G., Gentiloni N.: Paradoxical inhibition of cardiac lipid peroxidation in cancer patients treated with doxorubicin. J. Clin. Invest., 1996; 98: 650-661
[PubMed] [Full Text HTML] [Full Text PDF]
[38] Minotti G., Menna P., Salvatorelli E., Cairo G., Gianni L.: Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev., 2004; 56: 185-229
[PubMed] [Full Text HTML] [Full Text PDF]
[39] Murata T., Yamawaki H., Hori M., Sato K., Ozaki H., Karaki H.: Chronic vascular toxicity of doxorubicin in an organ cultured artery. Br. J. Pharmacol., 2001; 132: 1365-1373
[PubMed] [Full Text HTML] [Full Text PDF]
[40] Murata T., Yamawaki H., Yoshimoto R., Hori M., Sato K., Ozaki H., Karaki H.: Chronic effect of doxorubicin on vascular endothelium assesed by organ culture study. Life. Sci., 2001; 69: 2685-2695
[PubMed]
[41] Ng R., Better N., Green M.D.: Anticancer agents and cardiotoxicity. Semin. Oncol., 2006; 33: 2-14
[PubMed]
[42] Olson L.E., Bedja D., Alvey S.J., Cardounel A.J., Gabrielson K.L., Reeves R.H.: Protection from doxorubicin-induced cardiac toxicity in mice with a null allele of carbonyl reductase. Cancer Res., 2003; 63: 6602-6606
[PubMed] [Full Text HTML] [Full Text PDF]
[43] Pryor W.A.: Vitamin E and heart disease: basic science to clinical intervention trials. Free Radic. Biol. Med., 2000; 28: 141-164
[PubMed]
[44] Przybyszewski W.M.: Udział produktów peroksydacji lipidów w przeciwnowotworowym mechanizmie działania promieniowania jonizującego i cytostatyków radiomimetycznych. Post. Hig. Med. Dośw., 2001; 55: 803-813
[PubMed]
[45] Przybyszewski W.M., Widel M.: Activity of creatine kinase MB-isoenzyme in rat serum after heart irradiation and/or farmorubicin (4′-epidoxorubicin) treatment. Cancer Lett., 1996; 100: 145-150
[PubMed]
[46] Przybyszewski W.M., Widel M., Koterbicka A.: Early peroxidising effects of myocardial damage in rats after gamma-irradiation and farmorubicin (4′-epidoxorubicin) treatment. Cancer Lett., 1994; 81: 185-192
[PubMed]
[47] Przybyszewski W.M., Widel M., Koterbicka A.: Failure of cardioxane (ICRF-187, dexrazoxane) to limit peroxidative heart damage in rats after gamma irradiation or farmorubicin (4′-epidoxorubicin) treatment. Toxic Substance Mechanisms. 1997; 16: 133-149
[48] Przybyszewski W.M., Widel M., Koterbicka A.: Peroxidative changes in rat heart myocardium produced by gamma rays or farmorubicin: prevention by cardioxane or neoton. Toxic Substance Mechanisms. 1998; 17: 1-14
[49] Reif D.W., Schubert J., Aust S.D.: Iron release from feritin and lipid peroxidation by radiolytically generated reducing radicals. Arch. Biochem. Biophys., 1988; 264: 238-243
[PubMed]
[50] Saks V.A., Strumia E.: Phosphocreatine: Molecular and cellular aspects of the mechanism of cardioprotective action. Curr. Ther. Res., 1993; 53: 565-598
[51] Saraste A., Pulkki K., Kallajoki M., Heikkila P., Laine P., Mattila S., Nieminen M.S., Parvinen M., Voipio-Pulkki L.M.: Cardiomyocyte apoptosis and progression of heart failure to transplantation. Eur. J. Clin. Invest., 1999; 29: 380-386
[PubMed]
[52] Schultz-Hector S.: Radiation-induced heart disease. Int. J. Radiat. Biol., 1992; 61: 149-160
[PubMed]
[53] Schultz-Hector S., Sund M., Thames H.D.: Fractionation response and repair kinetics of radiation-induced heart failure in the rat. Radiother. Oncol., 1992; 23: 33-40
[PubMed]
[54] Stark G.: Functional consequences of oxidative membrane damage. J. Membr. Biol., 2005; 205: 1-16
[PubMed]
[55] Stark G.: The effect of ionizing radiation on lipid membranes. Biochem. Biophys. Acta, 1991; 1071: 103-122
[PubMed]
[56] Swain S.M., Whaley F.S., Ewer M.S.: Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer, 2003; 97: 2869-2879
[PubMed] [Full Text HTML] [Full Text PDF]
[57] Szuławska A., Czyż M.: Molekularne mechanizmy działania antracyklin. Post. Hig. Med. Dosw., 2006; 60: 78-100
[PubMed] [Full Text HTML] [Full Text PDF]
[58] Thomas C., Vile G.F., Winterbourn C.C.: The hydrolysis product of ICRF-187 promotes iron catalysed hydroxyl radical production via Fenton reaction. Biochem. Pharmacol., 1993; 45: 1967-1972
[PubMed]
[59] Vegesna V., Withers H.R., McBride W.H., Holly F.E.: Adriamycin-induced recall phenomenon after radiation pneumonitis and epilation in lung and hair follicles of mouse. Int. J. Radiat. Oncol. Biol. Phys., 1992; 23: 977-981
[PubMed]
[60] Weinstein D.M., Mihm M.J., Bauer J.A.: Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J. Pharmacol. Exp. Ther., 2000; 294: 396-401
[PubMed] [Full Text HTML] [Full Text PDF]
[61] Wojtacki J., Lewicka-Nowak E., Leśniewski-Kmak K.: Anthracycline-induced cardiotoxiocity: clinical course, risk factors, pathogenesis, detection and prevention-review of the literature. Med. Sci. Monit., 2000; 6: 411-420
[PubMed] [Full Text PDF]
[62] Wu S., Ko Y.S., Teng M.S., Ko Y.L., Hsu L.A., Hsueh C., Chou Y.Y., Liew C.C., Lee Y.S.: Adriamycin-induced cardiomyocyte and ebdothelial cell apoptosis: in vitro and in vivo studies. J. Mol. Cell Cardiol., 2002; 34: 1596-1607
[PubMed]
[63] Xu M.F., Tang P.L., Qian Z.M., Ashraf M.: Effects by doxorubicin on the myocardium are mediated by oxygen free radicals. Life Sci., 2001; 68: 889-901
[PubMed]
[64] Yamaoka M., Yamaguchi S., Suzuki T., Okuyama M., Nitobe J., Nakamura N., Mitsui Y., Tomoike H.: Apoptosis in rat cardiac myocytes induced by Fas ligand: priming for Fas-mediated apoptosis with doxorubicin. J. Mol. Cell Cardiol., 2000; 32: 881-889
[PubMed]
[65] Yeh E.T., Tong A.T., Lenihan D.J., Yusuf W., Swafford J., Champion C., Durand J.B., Gibbs H., Zafarmand A.A., Ewer M.S.: Cardiovascular complications of cancer therapy. Diagnosis, pathogenesis, and management. Circulation, 2004; 109: 3122-3131
[PubMed] [Full Text HTML] [Full Text PDF]
[66] Zhang J., Clark J.R.Jr., Herman E.H., Ferrans V.J.: Doxorubicin-induced apoptosis in spontaneously hypertensive rats: differential effects in heart, kidney and intestine and inhibition by ICRF-187. J. Mol. Cell Cardiol., 1996; 28: 1931-1943
[PubMed]
[67] Zucchi R., Danesi R.: Cardiac toxicity of antineoplastic anthracyclines. Curr. Med. Chem. Anticancer Agents, 2003; 3: 151-171
[PubMed]