
Iron metabolism and maternal-fetal iron circulation
Artur Słomka 1 , Ewa Żekanowska 1 , Katarzyna Piotrowska 2 , Justyna Kwapisz 1Streszczenie
Żelazo jest mikroelementem niezbędnym do prawidłowego funkcjonowania większości organizmów. Pierwiastek ten wykorzystywany jest w transporcie tlenu, syntezie mieliny i neuroprzekaźników, cyklach komórkowych oraz syntezie kwasów nukleinowych. Podkreśla się również wagę żelaza w procesie dojrzewania limfocytów i prawidłowym funkcjonowaniu narządu wzroku. Grupą szczególnie wrażliwą na zachwiania równowagi w gospodarce żelazem są noworodki. Niedobór żelaza w czasie ciąży niesie za sobą poważne konsekwencje kliniczne. Wydaje się, że główną rolę w transporcie żelaza między organizmem matki a płodu odgrywa hepcydyna oraz opisane w 2010 roku białko – zyklopen. W pracy przedstawiono fizjologiczną rolę białek uczestniczących w metabolizmie żelaza oraz opisano transport tego metalu przez łożysko.
Słowa kluczowe:żelazo • noworodki • niedobór żelaza • łożysko
Summary
Iron is an essential micronutrient for the proper functioning of most organisms. This element is used in oxygen transport, myelin and neurotransmitters synthesis, cell cycle and synthesis of nucleic acids. The importance of iron in the maturation of lymphocytes and proper functioning of the eye is also stressed. Newborns are particularly sensitive to imbalances in iron metabolism. Iron deficiency during pregnancy carries serious clinical consequences. It seems that a key role in the transport of iron between mother and fetus is plays by hepcidin and, described in 2010 zyklopen. The physiological role of proteins involved in iron metabolism and transport of this metal by the placenta are described in this paper.
Key words: iron • newborns • iron deficiency • placenta
Wykaz skrótów:
5-HT – 5-hydroksytryptamina (serotonina, 5-hydroxytryptamine); ACD – niedokrwistość chorób przewlekłych (anemia of chronic disease); apo-Tf – apo-transferryna (apo-transferrin); B2M – β2-mikroglobulina (β2-microglobulin); BeWo – linia ludzkich komórek łożyskowych (human placental choriocarcinoma cell line); BMPs – białka morfogenetyczne kości (bone morphogenetic proteins); C282Y – mutacja w genie HFE; CD228 – melanotransferryna (melanotransferrin); Cp – ceruloplazmina (ceruloplasmin); CytOx – oksydaza cytochromu c (cytochrome c oxidase); DCC – białko kodowane przez gen ulegający delecji w raku jelita grubego (deleted in colorectal carcinoma); Dcytb – cytochrom b dwunastnicy (duodenal cytochrome b); DFO – deferoksamina (desferrioxamine); DMT1/SLC11A2/Nramp2 – transporter metali dwuwartościowych (divalent metal transporter 1); FADS – desaturaza kwasów tłuszczowych (fatty acid desaturase); FNP/SLC40A1/IREG1/MTP-1 – ferroportyna (ferroportin); FNP/HFE – kompleks ferroportyny z białkiem HFE; GABA – kwas γ-aminomasłowy (gamma-aminobutyric acid); GABA-T – transaminaza GABA (4-aminobutyrate aminotransferase); GAD – dekarboksylaza kwasu glutaminowego (glutamic acid decarboxylase); GC – cyklaza guanylanowa (guanylate cyclase); GDF-15 – czynnik różnicowania wzrostu 15 (growth differentiation factor 15); Glu – kwas glutaminowy (glutamic acid); HAMP – gen kodujący hepcydynę (hepcidin antimicrobial peptide); HbE – hemoglobina E (hemoglobin E); HbH – hemoglobina H (hemoglobin H); HbS – hemoglobina S (hemoglobin S); Heph – hefajstyna (hephaestin); HFE – białko HFE (human hemochromatosis protein); HFE/B2M/TfR1 – kompleks białka HFE, β2-mikroglobuliny i receptora transferryny typu 1; HHCS – katarakta związana z hiperferrytynemią (hereditary hyperferritinemia-cataract syndrome); HIF – czynnik transkrypcji indukowany hipoksją (hypoxia-inducible factor 1); HJV/RGMc – hemojuwelina (hemojuvelin); IDA – niedokrwistość z niedoboru żelaza (iron-deficiency anemia); IGF-1 – insulinopodobny czynnik wzrostu 1 (insulin-like growth factor 1); IRE – sekwencje IRE (iron response element); IRIDA – niedokrwistość z niedoboru żelaza oporna na leczenie doustnymi preparatami żelaza (iron deficiency anemia refractory to oral iron therapy); IRP – białka regulujące gospodarkę żelazem (iron regulatory proteins); IUGR – hipotrofia wewnątrzmaciczna (intrauterine growth restriction); LEAP-1 – wątrobowy peptyd antybakteryjny 1 (hepcydyna, liver-expressed antimicrobial peptide); LVDCC – kanał dla wapnia (L-type voltage-dependent Ca2+ channel); MCF – ferrooksydaza miedziowa (multicopper ferroxidase); MHC – główny układ zgodności tkankowej (major histocompatibility complex); MPS – układ fagocytów jednojądrzastych (mononuclear phagocyte system); NEO – neogenina (neogenin); NGAL – lipokalina związana z żelatynazą neutrofilów (lipokalina-2, neutrophil gelatinase-associated lipocalin); Nramp – naturalne białko makrofagów związane z odpornością (natural resistance-associated macrophage protein); NTBI – żelazo niezwiązane z transferryną (non-transferrin bound iron); PHDs – hydroksylazy prolinowe (prolyl hydroxylases); RNR – reduktaza rybonukleotydowa (ribonucleotide reductase); RPE65 – 65 kDa izomerohydrolaza syntetyzowana przez nabłonek barwnikowy siatkówki (retinal pigment epithelium-specific 65 kDa protein); SCD – desaturaza stearoilo-CoA (stearoyl-CoA desaturase); sHJV – rozpuszczalna postać hemojuveliny (soluble hemojuvelin); STAT-3 – białka przekazujące sygnał i aktywujące transkrypcję (signal transducer and activator of transcription); sTfR – rozpuszczalny receptor transferryny (soluble transferrin receptor); SQR – oksydoreduktaza bursztynian-CoQ (succinate-coenzyme Q reductase); T3 – trijodotyronina (triiodothyronine); Tf(Fe)2 – transferryna (transferrin); TfR1/p90/CD71 – receptor transferryny typu 1 (transferrin receptor 1); Tf(Fe)2/TfR1 – kompleks żelazo-transferryna-receptor dla transferryny typu 1; TfR2 – receptor transferryny typu 2 (transferrin receptor 2); TGF-β – transformujący czynnik wzrostu beta (transforming growth factor beta); TMPRSS6 – matryptaza-2 (transmembrane protease, serine 6); TNF-α – czynnik martwicy nowotworów alfa (tumor necrosis factor alpha); TRH – tyreoliberyna (thyrotropin-releasing hormone); TTTS – zespół przetoczenia krwi między płodami (twin-to-twin transfusion syndrome); TWSG1 – białko TWSG1 (twisted gastrulation protein homolog 1); USF – czynnik transkrypcyjny (upstream stimulatory factor 1); UTR – sekwencje mRNA niepodlegające translacji (untranslated regions); VHL – białko supresorowe VHL (Von Hippel-Lindau tumor suppressor); Zp – zyklopen.
Rola żelaza w organizmie człowieka
Żelazo, ważny metal przejściowy w organizmie człowieka, odgrywa główną rolę w wielu procesach metabolicznych. Jest substratem w syntezie hemu, głównego składnika hemoglobiny, uczestniczy w reakcjach oksydo-redukcyjnych i immunologicznych oraz w syntezie DNA [31,57,62,109]. Średnia zawartość żelaza u dorosłego człowieka wynosi 4 g, co nie przekracza 0,01% masy ciała, z czego ponad 2 g znajdują się w hemoglobinie, 1 g zmagazynowany jest w hepatocytach, a pozostała część w białkach pełniących różnorodne funkcje [31,109].
Cechą charakterystyczną metabolizmu żelaza jest brak naturalnych mechanizmów odpowiedzialnych za usuwanie jego nadmiaru. Każdego dnia organizm traci 1-2 mg żelaza w wyniku złuszczania się nabłonka jelitowego, naskórka oraz krwawienia menstruacyjnego u kobiet. W przeciągu doby w wyniku fagocytozy zużytych erytrocytów makrofagi uwalniają około 20 mg żelaza, które wykorzystywane jest przede wszystkim w procesie erytropoezy, a niewielka ilość żelaza zostaje zmagazynowana w tkankach [31,43]. Zawartość żelaza w organizmie uzależniona jest głównie od „recyklingu” w układzie fagocytów jednojądrzastych (mononuclear phagocyte system, MPS), a żelazo wchłonięte w dwunastnicy nie przekracza 5% całkowitej zawartości tego pierwiastka w surowicy krwi [43].
Główną cechą decydującą o wytworzeniu precyzyjnych mechanizmów kontrolujących gospodarkę żelazem jest jego toksyczność. Żelazo może brać udział w reakcjach wolnorodnikowych, generując powstawanie rodnika hydroksylowego (HO•), reagującego ze wszystkimi biomolekułami, dlatego większość żelaza w organizmie człowieka związana jest z białkami [57,109]. Proteiny zawierające tę grupę prostetyczną podzielić można na cztery klasy: 1. Białka zawierające żelazo w postaci hemowej, niepełniące funkcji enzymatycznych – hemoglobina i mioglobina, odpowiedzialne za transport tlenu. 2. Białka zawierające centra żelazowo-siarkowe – 2Fe- 2S i 4Fe-4S – oksydoreduktaza dinukleotyd nikotynoamidoadeninowy-koenzym Q (NADH-coenzyme Q reductase), oksydoreduktaza bursztynian-CoQ (succinate-coenzyme Q reductase, SQR) i białko Rieskiego, które biorą udział w przenoszeniu elektronów podczas oddychania tlenowego, akonitaza mitochondrialna, biorąca udział w cyklu Krebsa oraz reduktaza rybonukleotydowa (ribonucleotide reductase, RNR), odpowiedzialna za syntezę i naprawę DNA. 3. Enzymy zawierające hem – kompleks cytochromu P-450, A, A3, B i C, odpowiedzialne za transport elektronów oraz katalaza, rozkładająca nadtlenek wodoru do tlenu i wody. 4. Białka zawierające żelazo w innej postaci – transferryna – Tf(Fe)2, laktoferryna (laktotransferryna) i mobilferryna, które transportują żelazo oraz ferrytyna i hemosyderyna, magazynujące żelazo [6,12,57,86,95,109].
Głównymi białkami uczestniczącymi w metabolizmie żelaza są: transferryna i jej receptory – TfR1 (transferrin receptor 1, p90, CD71), TfR2 (transferrin receptor 2) i rozpuszczalny receptor transferryny sTfR (soluble transferrin receptor), ferrytyna, transporter metali dwuwartościowych DMT1 (divalent metal transporter 1, solute carrier family 11 (proton-coupled divalent metal ion transporters), member 2, SLC11A2, natural resistance-associated macrophage protein 2, Nramp2), ferroportyna (ferroportin, FNP, solute carrier family 40 (iron-regulated transporter), member 1, SLC40A1, iron regulated-transporter-1, IREG1, metal transporter protein-1, MTP-1) oraz hepcydyna (liver-expressed antimicrobial peptide, LEAP-1). Poniżej przedstawiono fizjologiczną rolę tych białek w organizmie.
Transferryna
Do rodziny transferryn należą cztery białka: transferryna surowicza, owotransferryna (występująca u gadów i ptaków), laktoferryna i melanotransferryna (CD228) [61,82]. Transferryna jest białkiem o masie cząsteczkowej 79,6 kDa, zbudowanym z 679 aminokwasów, charakteryzującym się, w pH=7,4, bardzo silnym powinowactwem do jonów żelaza trójwartościowego (logKα=20,2) [101]. Transferryna syntetyzowana jest przede wszystkim w hepatocytach, chociaż jej wytwarzanie stwierdza się również w komórkach Sertoliego, oligodendrogleju i ependymy [9,23,24,51]. Obecność transferryny stwierdzono we krwi, limfie, płynie mózgowo-rdzeniowym, żółci, płynie owodniowym i mleku. Stężenie transferryny w surowicy w warunkach fizjologicznych wynosi 2,5 mg/ml, a wysycenie jej cząstek żelazem nie przekracza 30%. Główną funkcją transferryny jest transport żelaza z dwunastnicy i układu MPS do wszystkich komórek, a zwłaszcza do szpiku kostnego i komórek ulegających szybkim podziałom. W przebiegu niedokrwistości z niedoboru żelaza (iron-deficiency anemia, IDA) obserwuje się wzrost stężenia transferryny w surowicy krwi i wzrost ekspresji mRNA transferryny w hepatocytach. W stanach związanych z nadmierną kumulacją żelaza, w marskości wątroby i niedożywieniu stężenie transferryny jest obniżone. Melanotransferryna jest białkiem błonowym komórek czerniaka i tkanek płodu o niedokładnie poznanych funkcjach biologicznych. Białko to odgrywa niewielką rolę w transporcie żelaza, nasila natomiast proliferację komórek nowotworowych. Melanotransferryna znajdująca się na powierzchni komórek może również wiązać jony żelaza, zabezpieczając organizm przed ich udziałem w procesach Oksydo-redukcyjnych. Laktoferrynę zidentyfikowano w mleku, ślinie, łzach oraz ziarnistościach granulocytów wielojądrzastych. Białko to wykazuje działanie przeciwdrobnoustrojowe, głównie przez zdolność do chelatacji żelaza, niezbędnego do wzrostu mikroorganizmów. Laktoferryna uczestniczy także w absorpcji żelaza w jelicie oraz w odpowiedzi przeciwzapalnej. Podkreśla się także rolę tego białka w transporcie żelaza przez barierę krew-mózg (blood-brain barier, BBB) [61,82].
Receptor transferryny typu 1 (TfR1) i typu 2 (TfR2)
Receptor transferryny typu 1 należy do białek błonowych II typu, jest odpowiedzialny za wiązanie dwóch cząsteczek transferryny. Największą ekspresję TfR1 obserwuje się na powierzchni prekursorów erytrocytów, komórkach syncytiotrofoblastu i komórkach nowotworowych. Receptor TfR1 jest dimerem składającym się z dwóch identycznych podjednostek o masie 90 kDa każda, połączonych dwoma wiązaniami disiarczkowymi. Jego główną rolą w metabolizmie żelaza jest wiązanie dwóch cząsteczek transferryny, w wyniku czego następuje internalizacja kompleksu żelazo-transferryna-receptor (Tf(Fe)2/TfR1) i uwolnienie jonów żelaza do cytoplazmy komórek [1,47,61,82]. Do prawidłowego funkcjonowania receptora TfR1 niezbędne jest białko HFE (human hemochromatosis protein). Białko HFE zbudowane jest z 343 aminokwasów i należy do rodziny białek głównego układu zgodności tkankowej klasy I (major histocompatibility complex class I – MHC). Mutacja w genie HFE, polegająca na substytucji tyrozyny przez cysteinę w pozycji 282 łańcucha polipeptydowego (C282Y) jest najczęstszą przyczyną hemochromatozy. Białko HFE tworzy stabilny kompleks z receptorem transferryny typu 1, a następstwem połączenia tych dwóch białek jest zmniejszenie powinowactwa receptora do cząsteczek transferryny [25,50,79]. Ekspresja receptora transferryny typu 1 jest regulowana poprzez działanie dwóch białek IRP1 i IRP2, które współdziałają z sekwencjami IRE (iron responsive element), znajdującymi się w regionach 5′ lub 3′ mRNA TfR1 niepodlegających translacji (untranslated regions, UTR). W przypadku niedoboru żelaza białko IRP1 (w postaci apo-IRP1) przyłącza się do sekwencji IRE mRNA receptora transferryny typu 1 i chroni je przed degradacją, zwiększając syntezę TfR1. Natomiast nadmiar żelaza powoduje nabycie przez białko IRP1 aktywności enzymatycznej akonitazy, odpowiedzialnej za izomeryzację cytrynianu. W tej postaci (holo-IRP) białko to traci możliwość wiązania się z sekwencjami IRE, co w konsekwencji prowadzi do zmniejszenia syntezy TfR1 [47,57,82].
Receptor transferryny typu 2 opisali po raz pierwszy Kawabat i wsp. [45]. W wyniku ekspresji genu TfR2 umiejscowionego na chromosomie 7 (7q22) powstają dwa białka – TfR2a i TfR2b. TfR2a jest białkiem błonowym II typu i wykazuje dużą homologię z TfR1. W przeciwieństwie do TfR1, ekspresja receptora drugiego typu nie jest regulowana przez białka IRP, a wzrostu jego syntezy nie obserwuje się w niedoborze żelaza [29,45]. Głównym miejscem wytwarzania TfR2 są hepatocyty oraz erytroblasty [29,44]. Receptor transferryny typu 2 nie tworzy kompleksu z białkiem HFE, wykazuje również 25-krotnie mniejsze powinowactwo do cząsteczek transferryny w porównaniu do receptora pierwszego typu [110]. Dokładna rola TfR2 w metabolizmie żelaza nie została opisana, uważa się jednak, że receptor ten odpowiada za wiązanie żelaza niezwiązanego z transferryną (non-transferrin-bound iron, NTBI) [34].
Rozpuszczalny receptor transferryny (sTfR)
Postać rozpuszczalna receptora transferryny została opisana po raz pierwszy w 1986 roku przez Kohgo i wsp. jako polipeptyd o masie cząsteczkowej 84,9 kDa [46]. Dokładny mechanizm powstawania sTfR nie został opisany, ale przyjmuje się, że białko to pochodzi z dojrzewających erytrocytów, a w jego uwalnianie z eksosomów zaangażowane są proteazy granulocytów [39]. Liczba krążących cząsteczek sTfR jest proporcjonalna do liczby receptorów wbudowanych w błonę komórkową [83]. Oznaczanie stężenia sTfR jest szczególnie użyteczne w diagnozowaniu niedoboru żelaza u pacjentów ze stanem zapalnym. Rozpuszczalny receptor transferryny, w przeciwieństwie do ferrytyny, nie jest białkiem ostrej fazy, dlatego jego stężenie nie zależy od mediatorów stanu zapalnego [64]. Stężenie rozpuszczalnego receptora transferryny u zdrowych, dorosłych osób jest niezależne od płci, jednak wyższe stężenia tego białka obserwuje się u noworodków, dzieci i kobiet w ciąży, z najwyższymi wartościami w trzecim trymestrze ciąży [15,16]. Tabela 1 przedstawia kliniczną użyteczność oznaczania stężenia sTfR.
Tabela 1. Kliniczna użyteczność oznaczania stężenia rozpuszczalnego receptora transferryny (sTfR) [93]

Ferrytyna
Ferrytyna jest białkiem o masie cząsteczkowej 450 kDa, składającym się z 24 podjednostek mogących związać do 4500 atomów żelaza Fe3+. Białkowa część ferrytyny – apoferrytyna zbudowana jest z dwóch typów podjednostek – L (ferritin light chain) i H (ferritin heavy chain), kodowanych przez różne geny (L – 19q13.33; H – 11q13). Liczba podjednostek L przeważa w wątrobie i śledzionie, natomiast H w sercu i nerkach. Ferrytyna znajduje się przede wszystkim w cytoplazmie komórek, jej niewielkie ilości obserwuje się również w mitochondriach. Ferrytyna obecna w surowicy jest tylko niewielką frakcją ferrytyny komórkowej, jednak pula surowicza dobrze odzwierciedla zasoby żelaza w stanach fizjologii. Główną rolą ferrytyny jest magazynowanie jonów żelaza i zabezpieczanie tkanek przed jego toksycznym wpływem. Cząsteczki ferrytyny mają również aktywność enzymatyczną, utleniając jony Fe2+ do Fe3+ (głównie łańcuchy H, proces ten zachodzi 80 tysięcy razy szybciej niż w przypadku łańcuchów L). Podobnie jak synteza receptora transferryny typu 1, wytwarzanie ferrytyny kontrolowane jest przez białka IRP1 i IRP2. W razie niedoboru żelaza białka IRP przyłączają się do sekwencji IRE, hamując syntezę ferrytyny. Stężenie ferrytyny regulowane jest również przez cytokiny prozapalne. Czynnik martwicy guza TNF-α (tumor necrosis factor-alpha), interleukiny IL-1α i IL-6 odpowiedzialne są za wzrost syntezy ferrytyny H. Z kolei trijodotyronina (T3) i tyreoliberyna (thyrotropin-releasing hormone, TRH) odpowiedzialne za fosforylację białek IRP, insulina i insulinopodobny czynnik wzrostu IGF-1 (insulin-like growth factor 1) zwiększają syntezę podjednostek L i H ferrytyny [100,103]. Stanami, w których można obserwować wzrost stężenia ferrytyny są: nadmierna kumulacja żelaza, anemia w przebiegu chorób przewlekłych, zapalenie, infekcje, nowotwory i uszkodzenie wątroby. Zmniejszenie syntezy ferrytyny obserwuje się jedynie w niedoborze żelaza, jednak przy niskich wartościach stężenia tego białka (u kobiet 10 ng/ml, u mężczyzn 15 ng/ml) istotnie zmniejsza się czułość rozpoznania IDA (59%), przy bardzo dużej swoistości (99%) [58,64,99,100,103,104]. Mutacja w genie L ferrytyny może prowadzić do wystąpienia neuroferrytynopatii (dominant adult-onset basal ganglia disease) oraz katarakty związanej z hiperferrytynemią (hereditary hyperferritinemia-cataract syndrome – HHCS) [19,77].
Transporter metali dwuwartościowych (DMT-1, SLC11A2)
Białko DMT-1 po raz pierwszy opisali w 1997 roku Gunshin i wsp. [37]. Badacze ci stwierdzili, że DMT-1 jest 561-aminokwasowym białkiem należącym do rodziny Nramp (natural-resistance-associated macrophage protein) i jest przenośnikiem kationów dwuwartościowych: Fe2+, Zn2+, Mn2+, Co2+, Cd2+, Cu2+, Ni2+ i Pb2+ [37]. Ekspresję DMT-1 opisano na powierzchni wierzchołkowej enterocytów [37], na komórkach łożyska [33], neuronach istoty czarnej i jądra podstawnego [70] oraz w nerkach [26]. Ponadto, Fleming i wsp. wykazali, że transporter metali dwuwartościowych uczestniczy w transporcie żelaza z endosomu do cytoplazmy komórek [28]. DMT-1 jest niezbędny we wchłanianiu niehemowego żelaza w enterocytach i prekursorach erytrocytów [36]. Nie jest natomiast wymagany w magazynowaniu tego pierwiastka w wątrobie i jego transporcie przez łożysko [36]. Mutacja w genie DMT-1 odpowiedzialna jest za występowanie u pacjentów niedokrwistości mikrocytarnej z nadmierną kumulacją żelaza w hepatocytach, przy prawidłowym lub nieznacznie podwyższonym stężeniu ferrytyny w surowicy krwi [7]. Ze względu na to, że DMT-1 transportuje żelazo w postaci dwuwartościowej, przed rozpoczęciem tego procesu żelazo pokarmowe znajdujące się na trzecim stopniu utlenienia musi zostać zredukowane. W procesie tym uczestniczy zidentyfikowany przez McKie i wsp. cytochrom b dwunastnicy (duodenal cytochrome b, Dcytb) [67].
Hepcydyna i ferroportyna
Hepcydynę (wątrobowy peptyd antybakteryjny 1, liver-expressed antimicrobial peptide – LEAP-1) opisali po raz pierwszy Krause i wsp. w 2000 roku w filtratach krwi [48]. Rok później hepcydyn zidentyfikowali w moczu Park i wsp. [78]. Badacze ci stwierdzili, że hepcydyna jest syntetyzowana głównie w hepatocytach, występuje w trzech postaciach: 20-, 22- i 25-aminokwasowej oraz wykazuje działanie przeciwgrzybicze (Candida albicans, Aspergillus fumigatus, A. niger) oraz przeciwbakteryjne (Escherichia coli, Staphylococcus aureus, S. epidermidis, Streptococcus spp. grupy B) [78]. Fundamentalną rolę tego białka w metabolizmie żelaza potwierdzili Nicolas i wsp. W badaniach na myszach transgenicznych USF-/- (upstream stimulatory factor 2), będących laboratoryjnym modelem hemochromatozy z nokautem genu hepcydyny HAMP (hepcidin antimicrobial peptide), stwierdzili masywną kumulację żelaza w wątrobie, trzustce i sercu przy jednocześnie zmniejszonej zawartości żelaza w śledzionie [74]. Ta sama grupa naukowców w badaniach z wykorzystaniem transgenicznych myszy z nadekspresją genu HAMP stwierdziła ciężką niedokrwistość z niedoboru żelaza, co ostatecznie potwierdziło związek między hepcydyną i gospodarką żelazem [75].
Hepcydyna kodowana jest przez pojedynczy gen HAMP zawierający trzy eksony, zlokalizowane na długim ramieniu chromosomu 19 (19q13.1). Produktem ekspresji tego genu jest preprohepcydyna, 84-aminokwasowe białko zawierające 24-aminokwasowy peptyd sygnałowy. Preprohepcydyna ulega proteolitycznej modyfikacji polegającej na odcięciu peptydu sygnałowego, co skutkuje powstaniem 60-aminokwasowej prohepcydyny. Prohepcydyna jest następnie przekształcana przez konwertazę prohormonów – furynę do aktywnej, 25-aminokwasowej cząsteczki hepcydyny. Pozostałe dwie postaci hepcydyny – 20- i 22-aminokwasowa powstają prawdopodobnie wskutek rozkładu postaci 25-aminokwasowej lub podczas proteolitycznej obróbki [78,106].
W 2004 r. Nemeth i wsp. wyjaśnili, w jaki sposób hepcydyna reguluje gospodarkę żelazem. Zaobserwowali, że hepcydyna łączy się z ferroportyną (metal transporter protein, MTP-1), powodując jej internalizację i degradację, co skutkuje zmniejszeniem uwalniania żelaza z komórek do krwi [72]. Ferroportyna jest jedynym znanym białkowym eksporterem żelaza, występującym na powierzchni enterocytów, makrofagów, hepatocytów i komórek łożyska [72]. Stężenie ferroportyny jest regulowane przez niedobór żelaza i hipoksję, które odpowiedzialne są za wzrost jej syntezy oraz ostrą reakcję zapalną zmniejszającą jej wytwarzanie [21,68,92].
Synteza hepcydyny regulowana jest przez cztery podstawowe czynniki: zasoby żelaza w organizmie, stopień nasilenia erytropoezy, hipoksję i stan zapalny.
Regulacja ekspresji hepcydyny
Żelazo
Liczne prace eksperymentalne i kliniczne potwierdziły, że żelazo uczestniczy w regulacji syntezy hepcydyny, chociaż dokładny molekularny mechanizm tego procesu nie został wyjaśniony. Nicolas i wsp. wykazali, na modelach zwierzęcych, znaczny spadek stężenia mRNA hepcydyny w przebiegu anemii indukowanej flebotomiami i hemolizą [76]. Podobny stan obserwuje się u ludzi, w przebiegu niedokrwistości z niedoboru żelaza hepcydyna jest niewykrywalna lub jej stężenie jest bardzo niskie [32]. W procesie syntezy hepcydyny modulowanej przez żelazo uczestniczą białka morfogenetyczne kości (bone morphogenetic proteins, BMPs), należące do rodziny białek TGF-β (transforming growth factor-beta), a zwłaszcza BMP-6 [2]. Podawanie myszom dootrzewnowo białek BMP-6 powoduje wzrost ekspresji mRNA hepcydyny, co w konsekwencji prowadzi do zmniejszenia surowiczego stężenia żelaza i wysycenia transferryny [2]. Biologiczna aktywność BMP-6 zależy od białka hemojuweliny (HJV, repulsive guidance molecule c – RGMc) [2]. Hemojuwelina występuje w dwóch postaciach, jako białko błonowe oraz postać rozpuszczalna (soluble hemojuvelin – sHJV), która może być wykrywana w surowicy lub osoczu [56]. Hemojuwelina wbudowana w błonę komórkową, głównie hepatocytów, jest koreceptorem białek BMP, stymulującym ekspresję hepcydyny. Z kolei wstrzykiwanie myszom rozpuszczalnej HJV w postaci HJV.Fc (zewnątrzkomórkowa domena hemojuweliny połączona z fragmentem Fc ludzkiej immunoglobuliny klasy IgG) powodowało bezpośrednią interakcję HJV.Fc z BMP-6 i hamowało aktywność białek morfogenetycznych kości, prowadząc do zmniejszenia ekspresji hepcydyny, wzrostu ekspresji ferroportyny, a w konsekwencji wzrostu stężenia żelaza w surowicy krwi [2]. Również synteza hemojuweliny podlega kontroli molekularnej. W tym procesie uczestniczą dwa białka – neogenina (NEO), należąca do rodziny białek DCC (deleted in colorectal cancer) i matryptaza 2 (transmembrane protease, serine 6, TMPRSS6). Lee i wsp. wykazali, że neogenina stabilizuje błonową hemojuwelinę i tym samym hamuje sekrecję tego białka, zmniejszając ilość rozpuszczalnej HJV [52]. U myszy z nokautem genu NEO zaobserwowali nadmierną akumulację żelaza w hepatocytach, zmniejszoną ekspresję hepcydyny i zwiększoną ekspresję ferroportyny, co jednoznacznie przemawia za tym, że neogenina uczestniczy w regulacji homeostazy żelaza [52]. Matryptaza 2 odpowiada za rozszczepienie hemojuweliny znajdującej się na błonie komórkowej hepatocytów, co powoduje powstawanie nieaktywnych fragmentów HJV. Pozostawia jednak nienaruszoną postać rozpuszczalną tego białka, czego konsekwencją jest zmniejszenie syntezy hepcydyny [91].
Erytropoeza
Ashby i wsp. w 2010 r. wykazali, u zdrowych ochotników, spadek syntezy hepcydyny w wyniku wstrzyknięcia 5000 jednostek erytropoetyny (EPO) w postaci epoetyny β [3]. Autorzy podkreślili jednak, że zmniejszenie wytwarzania hepcydyny prawdopodobnie nie jest regulowane bezpośrednio przez cząsteczki EPO, ale w procesie tym mogą uczestniczyć szpikowe prekursory erytrocytów na różnym etapie dojrzewania [3]. Wśród czynników mogących hamować syntezę hepcydyny badacze zaproponowali: stężenie rozpuszczalnego receptora transferryny, wysycenie transferryny oraz czynnik różnicowania wzrostu GDF-15 (growth differentiation factor 15) [3]. GDF-15 jest białkiem należącym do rodziny TGF-β, którego wysoką ekspresję obserwuje się w czasie dojrzewania erytroblastów [97]. Wykazano, że duże stężenie czynnika GDF-15 odpowiedzialne jest za zmniejszenie syntezy hepcydyny [97]. Podobnie działa inne białko – TWSG1 (twisted gastrulation protein homolog 1) [98]. Proteina TWSG1 syntetyzowana jest przez erytroblasty przed rozpoczęciem procesu hemoglobinizacji i odpowiedzialna jest za zmniejszenie ekspresji hepcydyny poprzez interferencję z białkami BMP-2 i BMP-4 [98].
Hipoksja
Dokładny mechanizm supresji syntezy hepcydyny przez niedotlenienie nie został opisany, jednak przypuszcza się, że proces ten jest modulowany przez czynnik transkrypcji indukowany hipoksją HIF (hypoxia inducible transcription factor) oraz białko supresorowe VHL (von Hippel-Lindau tumor suppressor). HIF jest heterodimerem zawierającym trzy podjednostki regulatorowe – 1α, 2α i 3α. W obecności tlenu podjednostki regulatorowe ulegają modyfikacji przez zawierające żelazo hydroksylazy prolinowe (prolyl hydroxylases, PHDs), co prowadzi do ich ubikwitynacji i degradacji w proteasomie, w czym uczestniczy białko VHL. Hipoksja hamuje aktywność hydrolaz prolinowych, powodując akumulację podjednostek regulatorowych i ich przemieszczenie do jądra komórkowego. W tej sytuacji HIF-1α może wiązać się z promotorem genu hepcydyny, co z kolei hamuje jego transkrypcję i zmniejszanie syntezy hepcydyny. Niedobór żelaza powoduje wzrost syntezy HIF-1α w hepatocytach, a u myszy z nokautem genu HIF (HIF-1αflox/flox) karmionych dietą ubogożelazową obserwuje się znaczny wzrost syntezy hepcydyny [81].
Stan zapalny
Interleukina 6 (IL-6), główna cytokina prozapalna, odpowiedzialna jest za wzrost syntezy hepcydyny, podczas gdy IL-1 i czynnik martwicy nowotworów TNF-α nie uczestniczą w tym procesie [73]. IL-6 jest niezbędna do indukcji syntezy hepcydyny podczas stanu zapalnego, co w konsekwencji prowadzi do hipoferremii [71,112]. Połączenie IL-6 ze swoistym receptorem powoduje aktywację białek STAT-3 (signal transducer and activator of transcription 3), które przyłączają się do promotora genu hepcydyny, zwiększając jego ekspresję i tym samym syntezę białka [112]. Wyjaśnia to wysokie stężenie hepcydyny w stanach klinicznych przebiegających z reakcją zapalną, takich jak: anemia chorób przewlekłych (anemia of chronic disease, ACD), sepsa, czy nieswoiste zapalenie jelit (inflammatory bowel diseases) [92].
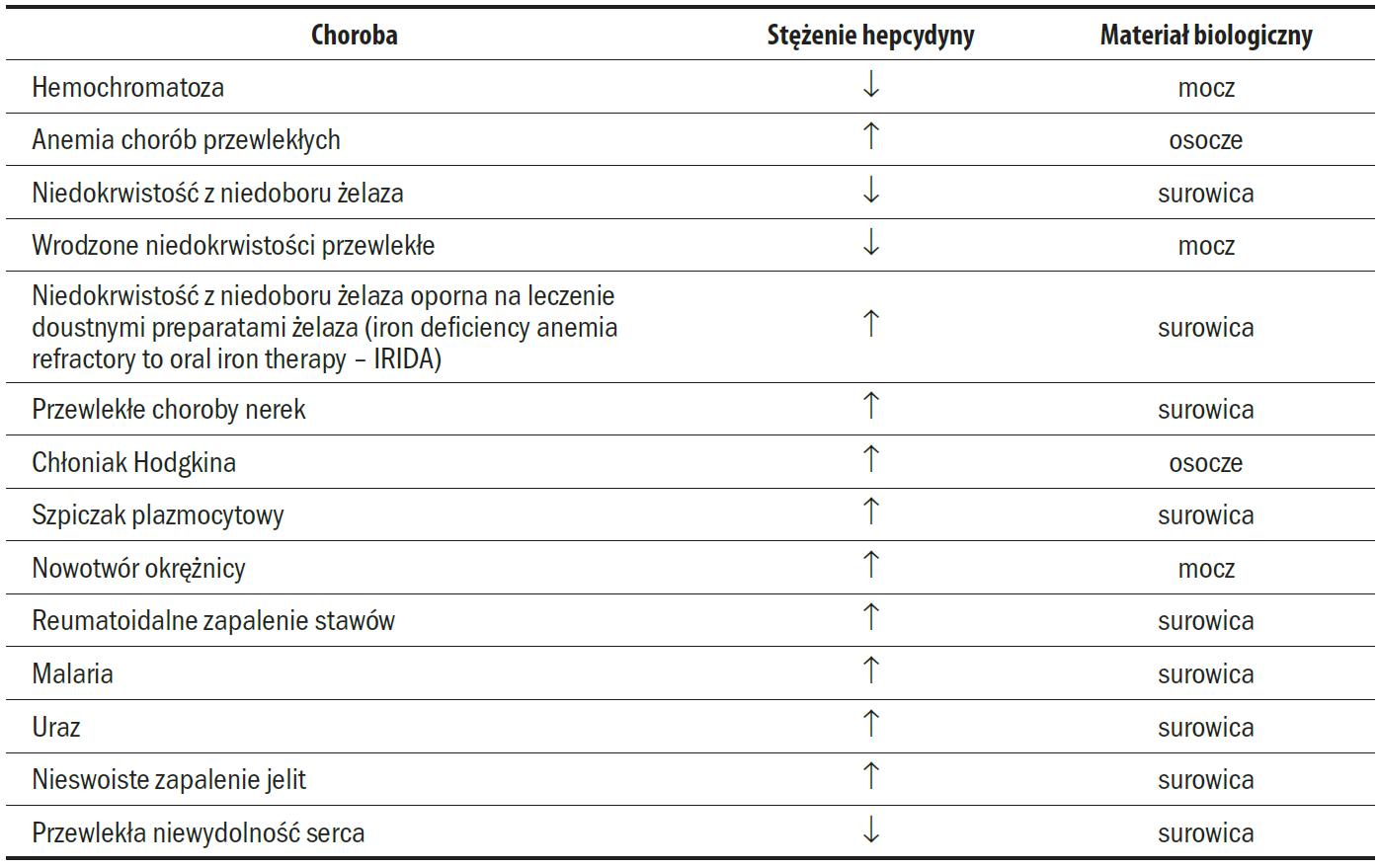
W tabeli 2 przedstawiono stężenie hepcydyny opisywane w różnych stanach klinicznych w porównaniu z grupą osób zdrowych.
Tabela 2. Stężenie hepcydyny w różnych stanach klinicznych w porównaniu do wartości obserwowanych w grupie osób zdrowych (strzałki – wzrost lub spadek stężenia) [92]
Udział żelaza w rozwoju ośrodkowego układu nerwowego
Żelazo uczestniczy przede wszystkim w przenoszeniu tlenu, jednak podkreśla się istotną rolę tego pierwiastka w rozwoju ośrodkowego układu nerwowego, w tym syntezy neuroprzekaźników i mieliny.
Żelazo bierze udział w tworzeniu osłonki mielinowej (rdzennej) aksonów, która niezbędna jest do prawidłowego przekazywania impulsów nerwowych [115]. Jeden atom żelaza jest składnikiem centrum aktywnego desaturazy stearoilo-CoA (stearoyl-CoA desaturase, SCD) [94], enzymu katalizującego syntezę oleinianu – prekursora kwasu nerwonowego [27]. Kwas nerwonowy jest z kolei głównym składnikiem cerebrozydów budujących osłonkę mielinową [40]. Niedobór żelaza w okresie prenatalnym może hamować syntezę kwasu nerwonowego, co w konsekwencji prowadzi do zaburzeń w mielinizacji aksonów [49,115]. Yu i wsp. zaobserwowali, że u 11-dniowych szczurów rasy Sprague Dawley z niedoborem żelaza (hematokryt 11,9%) zmniejsza się ilość mieliny w rdzeniu kręgowym, pniu mózgu i istocie białej móżdżku w porównaniu do grupy kontrolnej szczurów bez niedoboru żelaza (hematokryt 30,8%) [115]. Niedobór żelaza jest również przyczyną zmniejszonej aktywności oksydazy cytochromu c (cytochrome c oxidase – CytOx), enzymu zawierającego żelazo [105], który uczestniczy w fosforylacji oksydacyjnej, a jego aktywność odzwierciedla nasilenie procesów metabolicznych neuronów [20]. Deungria i wsp. stwierdzili, u szczurów rasy Sprague Dawley karmionych dietą ubogożelazową, zmniejszoną aktywność enzymu CytOx w obszarach mózgu odpowiedzialnych za wyższe funkcje życiowe, takich jak: hipokamp, kora gruszkowata, jądro przyśrodkowe wzgórza i kora zakrętu obręczy. Nie stwierdzili natomiast różnic w aktywności oksydazy cytochromu c w rejonach mózgu niezwiązanych z pamięcią: jądrze przednim wzgórza, jądrze bocznym ciała migdałowatego i jądrze przyśrodkowym uzdeczki. Na tej podstawie autorzy zasugerowali, że niedobór żelaza w czasie ciąży może prowadzić do zaburzeń procesów poznawczych, w tym uwagi i pamięci [20].
Żelazo jest również niezbędne do syntezy neuroprzekaźników w ośrodkowym układzie nerwowym. Wykazano, że utajony i wczesny niedobór żelaza powoduje zmniejszenie syntezy kwasu γ-aminomasłowego (gamma-aminobutyric acid – GABA) i kwasu glutaminowego (glutamic acid – Glu) wskutek zahamowania aktywności enzymów niezbędnych do ich syntezy [89,96]. Li wykazał, że u szczurów z niedokrwistością z niedoboru żelaza dochodzi do zmniejszenia aktywności dwóch enzymów uczestniczących w metabolizmie GABA: dekarboksylazy kwasu glutaminowego (glutamic acid decarboxylase, GAD) oraz transaminazy GABA (4-aminobutyrate aminotransferase, GABA-T) [53]. Duże ilości żelaza, niejednokrotnie przewyższające jego stężenie w hepatocytach, są umiejscowione w obszarach mózgu związanych z układem dopaminergicznym – gałce bladej, istocie czarnej, jądrze czerwiennym, ogoniastym i półleżącym oraz wzgórzu [114]. Niedobór żelaza prowadzi do zmniejszenia liczby receptorów dopaminergicznych D2, co w konsekwencji może się przyczynić do zmian w zachowaniu i zaburzeń w procesie uczenia się [114]. Jednocześnie ilość żelaza nie wpływa na syntezę monoamin czy aktywność enzymów biorących udział w ich metabolizmie [4]. Serotonina (5-hydroxytryptamine – 5-HT) jest kolejnym neuroprzekaźnikiem, którego biologiczna aktywność zależy od żelaza. Shukla i wsp. wykazali na modelach zwierzęcych, że w przebiegu utajonego niedoboru żelaza dochodzi do zaburzeń w syntezie serotoniny, bez możliwości cofnięcia tych zmian poprzez późniejszą suplementację żelazem [88].
Oprócz istotnej roli, jaką pełni żelazo w rozwoju centralnego układu nerwowego, pierwiastek ten jest również niezbędny do prawidłowego procesu widzenia. Funkcjonowanie fotoreceptorów w siatkówce jest uzależnione od zawierającego żelazo enzymu desaturazy kwasów tłuszczowych (fatty acid desaturase – FADS), a synteza cyklicznego guanozynomonofosforanu (cyclic guanosine monophosphate, cGMP), przekaźnika drugiego rzędu w procesie fototransdukcji, wymaga hemoproteiny – cyklazy guanylanowej (guanylate cyclase, GC) [87,113]. Moiseyev i wsp. wykazali, że izomerohydrolaza RPE65 (retinal pigment epithelium-specific 65 kDa protein) zawarta w nabłonku barwnikowym siatkówki, odpowiedzialna za izomeryzację estrów all-trans-retinylu do 11-cis retinolu, również wymaga żelaza do swojej prawidłowej aktywności [69].
Matczyno-płodowe krążenie żelaza
Całkowite zapotrzebowanie na żelazo u 55-kilogramowej kobiety w czasie ciąży wynosi około 1040 mg i zmienia się w kolejnych trymestrach ciąży [11]. W pierwszym trymestrze (1-3 miesiąc) zapotrzebowanie na żelazo spada ze względu na przerwę w miesiączkowaniu, co pozwala na zaoszczędzenie 160 mg żelaza [38]. Wzrost zapotrzebowania na ten pierwiastek obserwuje się od drugiego trymestru (4-6 miesiąc) i trwa do jej końca, ze szczególnym nasileniem w trzecim trymestrze (7-9 miesiąc), co związane jest ze wzrostem płodu [11]. Mechanizm transportu żelaza z organizmu matki do organizmu płodu nie jest dokładnie poznany. Zasadniczą rolę w matczyno-płodowym krążeniu żelaza odgrywa łożysko. Główną rolę w transporcie żelaza poprzez komórki syncytiotrofoblastu pełni receptor transferryny typu 1 [8]. Jego ekspresję obserwuje się przede wszystkim na powierzchni wierzchołkowej syncytiotrofoblastu [107]. Po związaniu dwóch cząsteczek transferryny kompleks żelazo-transferryna-receptor (Tf(Fe)2/TfR1) jest transportowany do cytoplazmy syncytiotrofoblastu w postaci endosomu [22]. Kwaśne środowisko panujące w endosomie, w utrzymanie którego zaangażowana jest H+-ATP-aza, powoduje uwolnienie żelaza z transferryny, podczas gdy białkowa część kompleksu Tf(Fe)2 – apo-transferryna (apo-Tf) pozostaje przyłączona do receptora TfR1 [65]. Do prawidłowego funkcjonowania receptora transferryny typu pierwszego niezbędna jest obecność białka HFE. Parkkila i wsp. wykazali ekspresję białka HFE na wierzchołkowej powierzchni komórek syncytiotrofoblastu [79]. Ponadto autorzy wykazali, że białko HFE jest połączone z β2-mikroglobuliną (β2-microglobulin, B2M) i taki kompleks łączy się z receptorem TfR. Kompleks białkowy HFE/B2M/TfR1 odgrywa główną rolę w transporcie żelaza przez łożysko, a mutacje w genie HFE mogą się przyczynić do nadmiernej kumulacji żelaza już w okresie prenatalnym [79]. Połączenie białka HFE z B2M stabilizuje proteinę HFE, umożliwia jej funkcjonowanie i prezentację na powierzchni komórek [108]. Waheed i wsp. stwierdzili, w hodowli komórek jajnika chomika chińskiego, że nadekspresja HFE bez nadekspresji B2M skutkuje zmniejszeniem komórkowej puli żelaza [108]. Nadekspresja obu białek – HFE i B2M prowadzi do wzrostu stężenia żelaza w cytoplazmie. Kompleks HFE-B2M nie wpływa na stopień powinowactwa receptora TfR1 do cząsteczek transferryny, jednak znacznie przyspiesza jego recykling i powoduje wzrost liczby cząsteczek TfR1 na powierzchni komórek łożyska [108].
Po uwolnieniu żelaza w endosomie apo-transferryna połączona z receptorem transferryny typu 1 ulega ponownemu przemieszczeniu na powierzchnię syncytiotrofoblastu, a cały proces zajmuje 7-10 min [66]. Żelazo zostaje uwolnione z endosomu do cytoplazmy za pomocą białka DMT1 [28,35]. Georgieff i wsp. stwierdzili, w tkankach łożyska, ekspresję białka DMT1 w cytoplazmie komórek syncytiotrofoblastu, na błonie podstawnej syncytiotrofoblastu i w komórkach Hofbauera, stanowiących rodzaj makrofagów i prawdopodobnie odpowiedzialnych za magazynowanie żelaza [33]. Nie zaobserwowali natomiast ekspresji DMT1 na powierzchni wierzchołkowej, co przemawia za tezą, że to białko odpowiada za transport żelaza z endosomu do cytoplazmy syncytiotrofoblastu [33]. Podobne wyniki otrzymali Chong i wsp. [17]. Stwierdzili oni również, że ekspresja DMT1 w łożysku jest zależna od żelaza, a liczba cząsteczek tego białka zwiększa się w niedoborze żelaza u matki [17]. Li i wsp. udowodnili, że dodatek silnego chelatora żelaza – deferroksaminy (desferrioxamine – DFO) do hodowli komórek łożyska BeWo (human placental choriocarcinoma cell line) powoduje wzrost ilości mRNA DMT1 oraz wzrost stężenia białka, z kolei dodatek apo-transferryny powoduje zmniejszenie ilości białka i jego mRNA [54]. Wyniki badań przeprowadzonych przez Gunshina i wsp. sugerują jednak, że DMT1 nie jest niezbędny do transportu żelaza z endosomu do cytoplazmy syncytiotrofoblastu [36]. U myszy z nokautem genu DMT1 (SLC11A2-/-) transport żelaza z organizmu matki do płodu był niezmieniony, dlatego też musi istnieć alternatywna droga takiego transportu. Autorzy zasugerowali, że w dodatkowym mechanizmie transportu żelaza może uczestniczyć lipokalina 2 (neutrophil gelatinase-associated lipocalin – NGAL) lub kanał dla wapnia LVDCC (L-type voltage-dependent Ca2+ channel) [36].
Kolejnym etapem w transporcie jonów żelaza do krwiobiegu płodu jest ich przejście przez błonę podstawnoboczną komórek łożyska, a w procesie tym uczestniczy opisana w 2000 r. ferroportyna [21,68]. Donovan i wsp. w badaniach na rybach z gatunku danio pręgowany (Danio rerio) stwierdzili, że FNP jest niezbędna w transporcie żelaza i wykazali jej ekspresję na błonie podstawnej ludzkiego łożyska [21]. Ekspresja ferroportyny w ludzkim łożysku nie ulega zmianie w przebiegu niedoboru żelaza w okresie ciąży [54,55]. Mao i wsp. wykazali, że mutacja typu null w genie ferroportyny u myszy powoduje obumarcie zarodków przed gastrulacją [63]. Z kolei u myszy z mutacją hipomorficzną obserwowano wady cewy nerwowej, takie jak: przepuklinę mózgową, rozszczep kręgosłupa i skrócenie przodomózgowia [63]. Z ferroportyną znajdującą się na powierzchni podstawnej syncytiotrofoblastu związane jest białko HFE, a ekspresję kompleksu FNP/HFE obserwuje się również w komórkach Hofbauera [5]. W cytoplazmie komórek łożyska dodatkowo zaobserwować można silną ekspresję ferrytyny, co przemawia za możliwością magazynowania żelaza przez syncytiotrofoblast [5].
Po opuszczeniu syncytiotrofoblastu jony żelaza muszą zostać utlenione do postaci trójwartościowej. Dokładny mechanizm tego procesu również nie jest opisany, ale najnowsze badania wskazują, że na tym etapie główną rolę może odgrywać zyklopen (Zp), białko opisane w 2010 roku przez Chena i wsp. [14]. Zyklopen jest ferrooksydazą miedziową (multicopper ferroxidase, MCF) należącą do tej samej rodziny białek co hefajstyna (hephaestin, Heph) i ceruloplazmina (ceruloplasmin, Cp) i wykazuje z nimi dużą homologię, jednak jej ekspresji nie obserwuje się w hepatocytach i enterocytach. Proteina ta jest odpowiedzialna za utlenienie jonów Fe2+ do Fe3+, które następnie łączą się z transferryną płodu [14].
Konsekwencje zaburzeń gospodarki żelazem w okresie prenatalnym
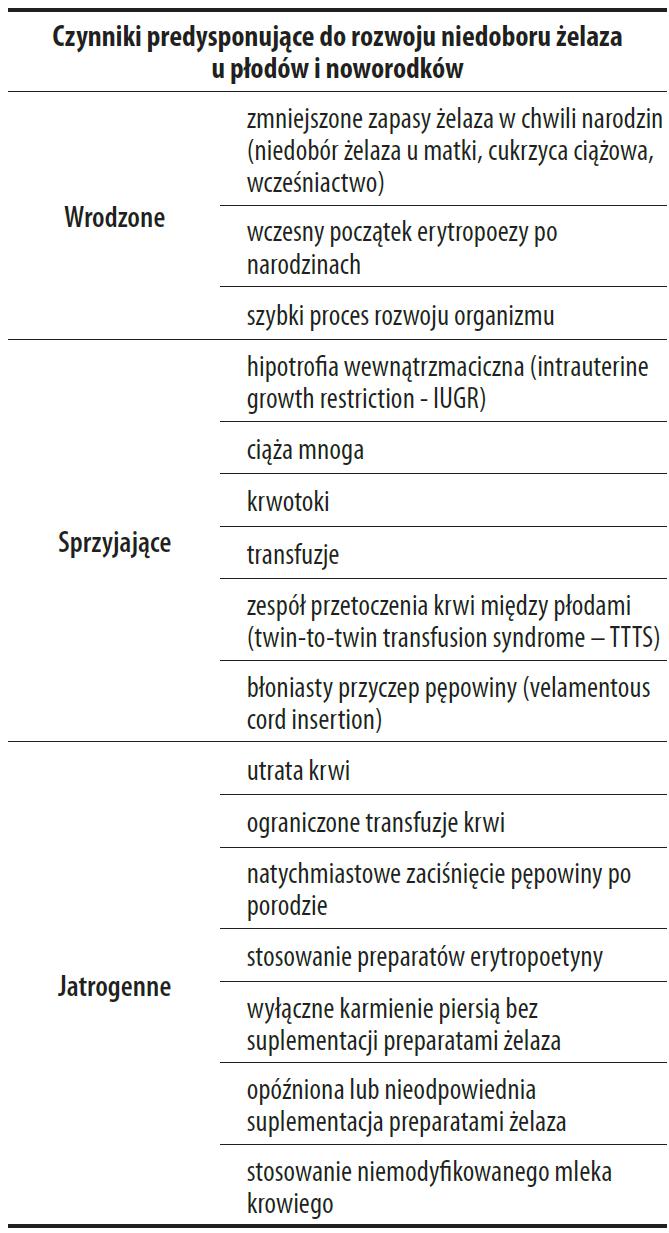
Status gospodarki żelazem u noworodka zależy od ilości żelaza dostarczonego z transferryną matki, dlatego zarówno niedobór, jak i nadmiar żelaza niosą za sobą poważne konsekwencje kliniczne, jednak niedokrwistość z niedoboru żelaza u matki nie zawsze jest przyczyną IDA u noworodków [90]. Zawartość żelaza u zdrowego płodu o masie 3500 g w trzecim trymestrze ciąży wynosi 7,5 mg/100 g masy ciała, natomiast u płodów urodzonych przedwcześnie o masie 2500 g i 1500 g wynosi odpowiednio: 7,4 mg i 7,1 mg/100 g masy ciała [111]. Tabela 3 przedstawia przyczyny niedoboru żelaza u płodów i noworodków.
Tabela 3. Przyczyny niedoboru żelaza u płodów i noworodków [84,85]
Główną konsekwencją niedoboru żelaza w okresie prenatalnym i noworodkowym są zaburzenia w budowie i funkcjonowaniu układu nerwowego. Zmniejszenie intensywności mielinizacji i syntezy neuroprzekaźników w życiu płodowym może skutkować: zmniejszeniem efektywności pamięci krótkotrwałej, zaburzeniami rozwoju poznawczego, dysfunkcją szlaków czołowo-prążkowiowych, większą nieśmiałością, słabszą orientacją i zaangażowaniem oraz zaburzeniami snu i słuchu w porównaniu do dzieci bez niedoboru żelaza [11,18,41,59,60,80]. Dziewiętnastoletnie obserwacje wykazały, że dzieci i młodzież, u których po urodzeniu rozpoznano niedobór żelaza, miały problemy z zachowaniem [18]. Jednocześnie niedobór żelaza nie wpływa na funkcję tarczycy i stężenie jej hormonów, jednak dzieci z niedoborem żelaza mogą mieć niewielkie zmiany w funkcjonowaniu osi podwzgórzowo-przysadkowej [102].
Podkreśla się również wpływ niedoboru żelaza w czasie ciąży na wskaźnik umieralności noworodków. Spadek stężenia hemoglobiny u matki poniżej 8 g/dl powoduje 2-3-krotny wzrost umieralności, podczas gdy spadek stężenia tego białka poniżej 5 g/dl powoduje 8-10-krotny wzrost ryzyka śmiertelności okołoporodowej noworodków [42]. Niedobór żelaza w czasie ciąży może skutkować wcześniactwem, mniejszą masą urodzeniową, rozwojem chorób układu krążenia, otyłości i cukrzycy typu 2 w późniejszych okresach życia [30]. Noworodki urodzone przez matki z niedoborem żelaza mają mniejsze wątrobowe zapasy żelaza, co świadczy o bezpośrednim wpływie niedoboru żelaza na rozwój prenatalny [30]. U matek ze zdiagnozowaną niedokrwistością z niedoboru żelaza ryzyko urodzenia wcześniaka jest 2,7 razy wyższe niż w przypadku zdrowych ciężarnych kobiet. Również ryzyko urodzenia noworodka z małą masa urodzeniową jest w przypadku IDA 3,1 razy wyższe. Jednocześnie noworodki urodzone przez matki z niedokrwistością syderopeniczną mają obniżoną odporność poprzez supresję limfocytów T [10].
PIŚMIENNICTWO
[1] Aisen P.: Transferrin receptor 1. Int. J. Biochem. Cell Biol., 2004; 36: 2137-2143
[PubMed]
[2] Andriopoulos B.Jr., Corradini E., Xia Y., Faasse S.A., Chen S., Grgurevic L., Knutson M.D., Pietrangelo A., Vukicevic S., Lin H.Y., Babitt J.L.: BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet., 2009; 41: 482-487
[PubMed]
[3] Ashby D.R., Gale D.P., Busbridge M., Murphy K.G., Duncan N.D., Cairns T.D., Taube D.H., Bloom S.R., Tam F.W., Chapman R., Maxwell P.H., Choi P.: Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin. Haematologica, 2010; 95: 505-508
[PubMed] [Full Text HTML] [Full Text PDF]
[4] Ashkenazi R., Ben-Shachar D., Youdim M.B.: Nutritional iron and dopamine binding sites in the rat brain. Pharmacol. Biochem. Behav., 1982; 17 (Suppl. 1): 43-47
[PubMed]
[5] Bastin J., Drakesmith H., Rees M., Sargent I., Townsend A.: Localisation of proteins of iron metabolism in the human placenta and liver. Br. J. Haematol., 2006; 134: 532-543
[PubMed] [Full Text HTML] [Full Text PDF]
[6] Beard J.L.: Iron biology in immune function, muscle metabolism and neuronal functioning. J. Nutr., 2001; 131 (Suppl. II): 568S-579S
[PubMed] [Full Text HTML] [Full Text PDF]
[7] Beaumont C., Delaunay J., Hetet G., Grandchamp B., de Montalembert M., Tchernia G.: Two new human DMT1 gene mutations in a patient with microcytic anemia, low ferritinemia, and liver iron overload. Blood, 2006; 107: 4168-4170
[PubMed] [Full Text HTML] [Full Text PDF]
[8] Bergamaschi G., Bergamaschi P., Carlevati S., Cazzola M.: Transferrin receptor expression in the human placenta. Haematologica, 1990; 75: 220-223
[PubMed]
[9] Beutler E., Gelbart T., Lee P., Trevino R., Fernandez M.A., Fairbanks V.F.: Molecular characterization of a case of atransferrinemia. Blood, 2000; 96: 4071-4074
[PubMed] [Full Text HTML] [Full Text PDF]
[10] Boccio J.R., Iyengar V.: Iron deficiency: causes, consequences, and strategies to overcome this nutritional problem. Biol. Trace Elem. Res., 2003; 94: 1-32
[PubMed]
[11] Bothwell T.H.: Iron requirements in pregnancy and strategies to meet them. Am. J. Clin. Nutr., 2000; 72 (Suppl. 1): 257S-264S
[PubMed] [Full Text HTML] [Full Text PDF]
[12] Brown E.N., Friemann R., Karlsson A., Parales J.V., Couture M.M., Eltis L.D., Ramaswamy S.: Determining Rieske cluster reduction potentials. J. Biol. Inorg. Chem., 2008; 13: 1301-1313
[PubMed]
[13] Carter R.C., Jacobson J.L., Burden M.J., Armony-Sivan R., Dodge N.C., Angelilli M.L., Lozoff B., Jacobson S.W.: Iron deficiency anemia and cognitive function in infancy. Pediatrics, 2010; 126: e427-e434
[PubMed] [Full Text HTML] [Full Text PDF]
[14] Chen H., Attieh Z.K., Syed B.A., Kuo Y.M., Stevens V., Fuqua B.K., Andersen H.S., Naylor C.E., Evans R.W., Gambling L., Danzeisen R., Bacouri-Haidar M., Usta J., Vulpe C.D., McArdle H.J.: Identification of zyklopen, a new member of the vertebrate multicopper ferroxidase family, and characterization in rodents and human cells. J. Nutr., 2010; 140: 1728-1735
[PubMed] [Full Text HTML] [Full Text PDF]
[15] Choi J.W., Im M.W., Pai S.H.: Serum transferrin receptor concentrations during normal pregnancy. Clin. Chem., 2000; 46: 725-727
[PubMed] [Full Text HTML] [Full Text PDF]
[16] Choi J.W., Pai S.H., Im M.W., Kim S.K.: Change in transferrin receptor concentrations with age. Clin. Chem., 1999; 45: 1562-1563
[PubMed] [Full Text HTML] [Full Text PDF]
[17] Chong W.S., Kwan P.C., Chan L.Y., Chiu P.Y., Cheung T.K., Lau T.K.: Expression of divalent metal transporter 1 (DMT1) isoforms in first trimester human placenta and embryonic tissues. Hum. Reprod., 2005; 20: 3532-3538
[PubMed] [Full Text HTML] [Full Text PDF]
[18] Corapci F., Calatroni A., Kaciroti N., Jimenez E., Lozoff B.: Longitudinal evaluation of externalizing and internalizing behavior problems following iron deficiency in infancy. J. Pediatr. Psychol., 2010; 35: 296-305
[PubMed] [Full Text HTML] [Full Text PDF]
[19] Curtis A.R., Fey C., Morris C.M., Bindoff L.A., Ince P.G., Chinnery P.F., Coulthard A., Jackson M.J., Jackson A.P., McHale D.P., Hay D., Barker W.A., Markham A.F., Bates D., Curtis A., Burn J.: Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat. Genet., 2001; 28: 350-354
[PubMed]
[20] de Deungria M., Rao R., Wobken J.D., Luciana M., Nelson C.A., Georgieff M.K.: Perinatal iron deficiency decreases cytochrome c oxidase (CytOx) activity in selected regions of neonatal rat brain. Pediatr. Res., 2000; 48: 169-176
[PubMed] [Full Text HTML] [Full Text PDF]
[21] Donovan A., Brownlie A., Zhou Y., Shepard J., Pratt S.J., Moynihan J., Paw B.H., Drejer A., Barut B., Zapata A., Law T.C., Brugnara C., Lux S.E., Pinkus G.S., Pinkus J.L., Kingsley P.D., Palis J., Fleming M.D., Andrews N.C., Zon L.I.: Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature, 2000; 403: 776-781
[PubMed]
[22] Douglas G.C., King B.F.: Uptake and processing of 125I-labelled transferrin and 59Fe-labelled transferrin by isolated human trophoblast cells. Placenta, 1990; 11: 41-57
[PubMed]
[23] Espinosa de los Monteros A., Kumar S., Scully S., Cole R., de Vellis J.: Transferrin gene expression and secretion by rat brain cells in vitro. J. Neurosci. Res., 1990; 25: 576-580
[PubMed]
[24] Esterle T.M., Sanders-Bush E.: Serotonin agonists increase transferrin levels via activation of 5-HT1C receptors in choroid plexus epithelium. J. Neurosci., 1992; 12: 4775-4782
[PubMed] [Full Text PDF]
[25] Feder J.N., Penny D.M., Irrinki A., Lee V.K., Lebrón J.A., Watson N., Tsuchihashi Z., Sigal E., Bjorkman P.J., Schatzman R.C.: The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc. Natl. Acad. Sci. USA, 1998; 95: 1472-1477
[PubMed] [Full Text HTML] [Full Text PDF]
[26] Ferguson C.J., Wareing M., Ward D.T., Green R., Smith C.P., Riccardi D.: Cellular localization of divalent metal transporter DMT-1 in rat kidney. Am. J. Physiol. Renal Physiol., 2001; 280: F803-F814
[PubMed] [Full Text HTML] [Full Text PDF]
[27] Fewster M.E., Ihrig T., Mead J.F.: Biosynthesis of long chain fatty acids by oligodendroglia isolated from bovine white matter. J. Neurochem., 1975; 25: 207-213
[PubMed]
[28] Fleming M.D., Romano M.A., Su M.A., Garrick L.M., Garrick M.D., Andrews N.C.: Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc. Natl. Acad. Sci. USA, 1998; 95: 1148-1153
[PubMed] [Full Text HTML] [Full Text PDF]
[29] Fleming R.E., Migas M.C., Holden C.C., Waheed A., Britton R.S., Tomatsu S., Bacon B.R., Sly W.S.: Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA, 2000; 97: 2214-2219
[PubMed] [Full Text HTML] [Full Text PDF]
[30] Gambling L., Kennedy C., McArdle H.J.: Iron and copper in fetal development. Semin. Cell Dev. Biol., 2011; 22: 637-644
[PubMed]
[31] Ganz T.: Molecular control of iron transport. J. Am. Soc. Nephrol., 2007; 18: 394-400
[PubMed] [Full Text HTML] [Full Text PDF]
[32] Ganz T., Olbina G., Girelli D., Nemeth E., Westerman M.: Immunoassay for human serum hepcidin. Blood, 2008; 112: 4292-4297
[PubMed] [Full Text HTML] [Full Text PDF]
[33] Georgieff M.K., Wobken J.K., Welle J., Burdo J.R., Connor J.R.: Identification and localization of divalent metal transporter-1 (DMT-1) in term human placenta. Placenta, 2000; 21: 799-804
[PubMed]
[34] Graham R.M., Reutens G.M., Herbison C.E., Delima R.D., Chua A.C., Olynyk J.K., Trinder D.: Transferrin receptor 2 mediates uptake of transferrin-bound and non-transferrin-bound iron. J. Hepatol., 2008; 48: 327-334
[PubMed]
[35] Gruper Y., Bar J., Bacharach E., Ehrlich R.: Transferrin receptor co-localizes and interacts with the hemochromatosis factor (HFE) and the divalent metal transporter-1 (DMT1) in trophoblast cells. J. Cell. Physiol., 2005; 204: 901-912
[PubMed]
[36] Gunshin H., Fujiwara Y., Custodio A.O., Direnzo C., Robine S., Andrews N.C.: Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J. Clin. Invest., 2005; 115: 1258-1266
[PubMed] [Full Text HTML] [Full Text PDF]
[37] Gunshin H., Mackenzie B., Berger U.V., Gunshin Y., Romero M.F., Boron W.F., Nussberger S., Gollan J.L., Hediger M.A.: Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature, 1997; 388: 482-488
[PubMed]
[38] Hallberg L., Rossander-Hultén L.: Iron requirements in menstruating women. Am. J. Clin. Nutr., 1991; 54: 1047-1058
[PubMed] [Full Text PDF]
[39] Johnstone R.M.: Cleavage of the transferrin receptor by human granulocytes: differential proteolysis of the exosome-bound TFR. J. Cell. Physiol., 1996; 168: 333-345
[PubMed]
[40] Joseph K.C., Druse M.J., Newell L.R., Hogan E.L.: Fatty acid composition of cerebrosides, sulphatides and ceramides in murine leucodystrophy: the quaking mutant. J. Neurochem., 1972; 19: 307-312
[PubMed]
[41] Jougleux J.L., Rioux F.M., Church M.W., Fiset S., Surette M.E.: Mild maternal iron deficiency anemia during pregnancy and lactation in guinea pigs causes abnormal auditory function in the offspring. J. Nutr., 2011; 141: 1390-1395
[PubMed] [Full Text HTML] [Full Text PDF]
[42] Kalaivani K.: Prevalence & consequences of anaemia in pregnancy. Indian J. Med. Res., 2009; 130: 627-633
[PubMed] [Full Text PDF]
[43] Kaplan J., Ward D.M., De Domenico I.: The molecular basis of iron overload disorders and iron-linked anemias. Int. J. Hematol., 2011; 93: 14-20
[PubMed]
[44] Kawabata H., Nakamaki T., Ikonomi P., Smith R.D., Germain R.S., Koeffler H.P.: Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood, 2001; 98: 2714-2719
[PubMed] [Full Text PDF]
[45] Kawabata H., Yang R., Hirama T., Vuong P.T., Kawano S., Gombart A.F., Koeffler H.P.: Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem., 1999; 274: 20826-20832
[PubMed] [Full Text HTML] [Full Text PDF]
[46] Kohgo Y., Nishisato T., Kondo H., Tsushima N., Niitsu Y., Urushizaki I.: Circulating transferrin receptor in human serum. Br. J. Haematol., 1986; 64: 277-281
[PubMed]
[47] Kohgo Y., Torimoto Y., Kato J.: Transferrin receptor in tissue and serum: updated clinical significance of soluble receptor. Int. J. Hematol., 2002; 76: 213-218
[PubMed]
[48] Krause A., Neitz S., Mägert H.J., Schulz A., Forssmann W.G., Schulz-Knappe P., Adermann K.: LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett., 2000; 480: 147-150
[PubMed]
[49] Larkin E.C., Jarratt B.A., Rao G.A.: Reduction of relative levels of nervonic to lignoceric acid in the brain of rat pups due to iron deficiency. Nutr. Res., 1986; 6: 309-317
[Abstract]
[50] Lebrón J.A., Bennett M.J., Vaughn D.E., Chirino A.J., Snow P.M., Mintier G.A., Feder J.N., Bjorkman P.J.: Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell, 1998; 93: 111-123
[PubMed] [Full Text HTML]
[51] Lécureuil C., Saleh M.C., Fontaine I., Baron B., Zakin M.M., Guillou F.: Transgenic mice as a model to study the regulation of human transferrin expression in Sertoli cells. Hum. Reprod., 2004; 19: 1300-1307
[PubMed] [Full Text HTML] [Full Text PDF]
[52] Lee D.H., Zhou L.J., Zhou Z., Xie J.X., Jung J.U., Liu Y., Xi C.X., Mei L., Xiong W.C.: Neogenin inhibits HJV secretion and regulates BMP-induced hepcidin expression and iron homeostasis. Blood, 2010; 115: 3136-3145
[PubMed] [Full Text HTML] [Full Text PDF]
[53] Li D.: Effects of iron deficiency on iron distribution and gamma-aminobutyric acid (GABA) metabolism in young rat brain tissues. Hokkaido Igaku Zasshi, 1998; 73: 215-225
[PubMed]
[54] Li Y.Q., Bai B., Cao X.X., Zhang Y.H., Yan H., Zheng Q.Q., Zhuang G.H.: Divalent metal transporter 1 expression and regulation in human placenta. Biol. Trace Elem. Res., 2012; 146: 6-12
[PubMed]
[55] Li Y.Q., Yan H., Bai B.: Change in iron transporter expression in human term placenta with different maternal iron status. Eur. J. Obstet. Gynecol. Reprod. Biol., 2008; 140: 48-54
[PubMed]
[56] Lin L., Goldberg Y.P., Ganz T.: Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood, 2005; 106: 2884-2889
[PubMed] [Full Text HTML] [Full Text PDF]
[57] Lipiński P., Starzyński R.R.: Rola białek IRP (iron regulatory proteins) w regulacji ogólnoustrojowej homeostazy żelaza: lekcje płynące z badań na myszach z nokautem genów Irp1 i Irp2. Postępy Hig. Med. Dośw., 2006; 60: 322-330
[PubMed] [Full Text HTML] [Full Text PDF]
[58] Liu X., Theil E.C.: Ferritins: dynamic management of biological iron and oxygen chemistry. Acc. Chem. Res., 2005; 38: 167-175
[PubMed]
[59] Lozoff B., Clark K.M., Jing Y., Armony-Sivan R., Angelilli M.L., Jacobson S.W.: Dose-response relationships between iron deficiency with or without anemia and infant social-emotional behavior. J. Pediatr., 2008; 152: 696-702
[PubMed] [Full Text HTML] [Full Text PDF]
[60] Lukowski A.F., Koss M., Burden M.J., Jonides J., Nelson C.A., Kaciroti N., Jimenez E., Lozoff B.: Iron deficiency in infancy and neurocognitive functioning at 19 years: evidence of long-term deficits in executive function and recognition memory. Nutr. Neurosci., 2010; 13: 54-70
[PubMed] [Full Text PDF]
[61] Macedo M.F., de Sousa M.: Transferrin and the transferrin receptor: of magic bullets and other concerns. Inflamm. Allergy Drug Targets, 2008; 7: 41-52
[PubMed]
[62] Małyszko J.: Hepcidin assays: ironing out some details. Clin. J. Am. Soc. Nephrol., 2009; 4: 1015-1016
[PubMed] [Full Text HTML] [Full Text PDF]
[63] Mao J., McKean D.M., Warrier S., Corbin J.G., Niswander L., Zohn I.E.: The iron exporter ferroportin 1 is essential for development of the mouse embryo, forebrain patterning and neural tube closure. Development, 2010; 137: 3079-3088
[PubMed] [Full Text HTML] [Full Text PDF]
[64] Mast A.E., Blinder M.A., Gronowski A.M., Chumley C., Scott M.G.: Clinical utility of the soluble transferrin receptor and comparison with serum ferritin in several populations. Clin. Chem., 1998; 44: 45-51
[PubMed] [Full Text HTML] [Full Text PDF]
[65] McArdle H.J., Douglas A.J., Bowen B.J., Morgan E.H.: The mechanism of iron uptake by the rat placenta. J. Cell. Physiol., 1985; 124: 446-450
[PubMed]
[66] McArdle H.J., Douglas A.J., Morgan E.H.: Uptake of transferrin and iron by cultured rat placental cells. J. Cell. Physiol., 1985; 122: 405-409
[PubMed]
[67] McKie A.T., Barrow D., Latunde-Dada G.O., Rolfs A., Sager G., Mudaly E., Mudaly M., Richardson C., Barlow D., Bomford A., Peters T.J., Raja K.B., Shirali S., Hediger M.A., Farzaneh F., Simpson R.J.: An iron-regulated ferric reductase associated with the absorption of dietary iron. Science, 2001; 291: 1755-1759
[PubMed]
[68] McKie A.T., Marciani P., Rolfs A., Brennan K., Wehr K., Barrow D., Miret S., Bomford A., Peters T.J., Farzaneh F., Hediger M.A., Hentze M.W., Simpson R.J.: A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell, 2000; 5: 299-309
[PubMed] [Full Text HTML] [Full Text PDF]
[69] Moiseyev G., Chen Y., Takahashi Y., Wu B.X., Ma J.X.: RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc. Natl. Acad. Sci. USA, 2005; 102: 12413-12418
[PubMed] [Full Text HTML] [Full Text PDF]
[70] Moos T., Trinder D., Morgan E.H.: Cellular distribution of ferric iron, ferritin, transferrin and divalent metal transporter 1 (DMT1) in substantia nigra and basal ganglia of normal and beta2-microglobulin deficient mouse brain. Cell. Mol. Biol., 2000; 46: 549-561
[PubMed]
[71] Nemeth E., Rivera S., Gabayan V., Keller C., Taudorf S., Pedersen B.K., Ganz T.: IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Invest., 2004; 113: 1271-1276
[PubMed] [Full Text HTML] [Full Text PDF]
[72] Nemeth E., Tuttle M.S., Powelson J., Vaughn M.B., Donovan A., Ward D.M., Ganz T., Kaplan J.: Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science, 2004; 306: 2090-2093
[PubMed]
[73] Nemeth E., Valore E.V., Territo M., Schiller G., Lichtenstein A., Ganz T.: Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood, 2003; 101: 2461-2463
[PubMed] [Full Text HTML] [Full Text PDF]
[74] Nicolas G., Bennoun M., Devaux I., Beaumont C., Grandchamp B., Kahn A., Vaulont S.: Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA, 2001; 98: 8780-8785
[PubMed] [Full Text HTML] [Full Text PDF]
[75] Nicolas G., Bennoun M., Porteu A., Mativet S., Beaumont C., Grandchamp B., Sirito M., Sawadogo M., Kahn A., Vaulont S.: Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. USA, 2002; 99: 4596-4601
[PubMed] [Full Text HTML] [Full Text PDF]
[76] Nicolas G., Chauvet C., Viatte L., Danan J.L., Bigard X., Devaux I., Beaumont C., Kahn A., Vaulont S.: The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest., 2002; 110: 1037-1044
[PubMed] [Full Text HTML] [Full Text PDF]
[77] Papanikolaou G., Chandrinou H., Bouzas E., Contopoulos-Ioannidis D., Kalotychou V., Prentzas K., Lilakos K., Asproudis I., Palaiologou D., Premetis E., Papassotiriou I., Sakellaropoulos N.: Hereditary hyperferritinemia cataract syndrome in three unrelated families of western Greek origin caused by the C39 > G mutation of L-ferritin IRE. Blood Cells Mol. Dis., 2006; 36: 33-40
[PubMed]
[78] Park C.H., Valore E.V., Waring A.J., Ganz T.: Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem., 2001; 276: 7806-7810
[PubMed] [Full Text HTML] [Full Text PDF]
[79] Parkkila S., Waheed A., Britton R.S., Bacon B.R., Zhou X.Y., Tomatsu S., Fleming R.E., Sly W.S.: Association of the transferrin receptor in human placenta with HFE, the protein defective in hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA, 1997; 94: 13198-13202
[PubMed] [Full Text HTML] [Full Text PDF]
[80] Peirano P.D., Algarín C.R., Garrido M.I., Lozoff B.: Iron deficiency anemia in infancy is associated with altered temporal organization of sleep states in childhood. Pediatr. Res., 2007; 62: 715-719
[PubMed] [Full Text HTML] [Full Text PDF]
[81] Peyssonnaux C., Zinkernagel A.S., Schuepbach R.A., Rankin E., Vaulont S., Haase V.H., Nizet V., Johnson R.S.: Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Invest., 2007; 117: 1926-1932
[PubMed] [Full Text HTML] [Full Text PDF]
[82] Qian Z.M., Li H., Sun H., Ho K.: Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol. Rev., 2002; 54: 561-587
[PubMed] [Full Text HTML] [Full Text PDF]
[83] R’zik S., Beguin Y.: Serum soluble transferrin receptor concentration is an accurate estimate of the mass of tissue receptors. Exp. Hematol., 2001; 29: 677-685
[PubMed]
[84] Rao R., Georgieff M.K.: Iron in fetal and neonatal nutrition. Semin. Fetal Neonatal Med., 2007; 12: 54-63
[PubMed]
[85] Rao R., Georgieff M.K.: Perinatal aspects of iron metabolism. Acta Paediatr. Suppl., 2002; 91: 124-129
[PubMed]
[86] Sadrzadeh S.M., Saffari Y.: Iron and brain disorders. Am. J. Clin. Pathol., 2004; 121 (Suppl.): S64-S70
[PubMed]
[87] Shichi H.: Microsomal electron transfer system of bovine retinal pigment epithelium. Exp. Eye Res., 1969; 8: 60-68
[PubMed]
[88] Shukla A., Agarwal K.N., Chansuria J.P., Taneja V.: Effect of latent iron deficiency on 5-hydroxytryptamine metabolism in rat brain. J. Neurochem., 1989; 52: 730-735
[PubMed]
[89] Shukla A., Agarwal K.N., Shukla G.S.: Latent iron deficiency alters gamma-aminobutyric acid and glutamate metabolism in rat brain. Experientia, 1989; 45: 343-345
[PubMed]
[90] Sichieri R., Fonseca V.M., Hoffman D., Trugo N.M., Moura A.S.: Lack of association between iron status at birth and growth of preterm infants. Rev. Saude Publica, 2006; 40: 641-647
[PubMed] [Full Text PDF]
[91] Silvestri L., Pagani A., Nai A., De Domenico I., Kaplan J., Camaschella C.: The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab., 2008; 8: 502-511
[PubMed] [Full Text HTML] [Full Text PDF]
[92] Słomka A., Koba M., Kulwas A., Żekanowska E.: Hepcidin: biological activity, analytical methods in biological fluids, clinical applications and antagonists. A short review. Curr. Pharm. Anal., 2011; 7: 160-166
[Abstract]
[93] Speeckaert M.M., Speeckaert R., Delanghe J.R.: Biological and clinical aspects of soluble transferrin receptor. Crit. Rev. Clin. Lab. Sci., 2010; 47: 213-228
[Abstract]
[94] Strittmatter P., Spatz L., Corcoran D., Rogers M.J., Setlow B., Redline R.: Purification and properties of rat liver microsomal stearyl coenzyme A desaturase. Proc. Natl. Acad. Sci. USA, 1974; 71: 4565-4569
[PubMed] [Full Text PDF]
[95] Tamarit J., Mulliez E., Meier C., Trautwein A., Fontecave M.: The anaerobic ribonucleotide reductase from Escherichia coli. The small protein is an activating enzyme containing a [4fe-4s]2+ center. J. Biol. Chem., 1999; 274: 31291-31296
[PubMed] [Full Text HTML] [Full Text PDF]
[96] Taneja V., Mishra K., Agarwal K.N.: Effect of early iron deficiency in rat on the γamma-aminobutyric acid shunt in brain. J. Neurochem., 1986; 46: 1670-1674
[PubMed]
[97] Tanno T., Bhanu N.V., Oneal P.A., Goh S.H., Staker P., Lee Y.T., Moroney J.W., Reed C.H., Luban N.L., Wang R.H., Eling T.E., Childs R., Ganz T., Leitman S.F., Fucharoen S., Miller J.L.: High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med., 2007; 13: 1096-1101
[PubMed]
[98] Tanno T., Porayette P., Sripichai O., Noh S.J., Byrnes C., Bhupatiraju A., Lee Y.T., Goodnough J.B., Harandi O., Ganz T., Paulson R.F., Miller J.L.: Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood, 2009; 114: 181-186
[PubMed] [Full Text HTML] [Full Text PDF]
[99] Theil E.C.: Ferritin iron minerals are chelator targets, antioxidants, and coated, dietary iron. Ann. N.Y. Acad. Sci., 2010; 1202: 197-204
[PubMed]
[100] Thomson A.M., Rogers J.T., Leedman P.J.: Iron-regulatory proteins, iron-responsive elements and ferritin mRNA translation. Int. J. Biochem. Cell Biol., 1999; 31: 1139-1152
[PubMed]
[101] Thorstensen K., Romslo I.: The role of transferrin in the mechanism of cellular iron uptake. Biochem. J., 1990; 271: 1-9
[PubMed] [Full Text PDF]
[102] Tienboon P., Unachak K.: Iron deficiency anaemia in childhood and thyroid function. Asia Pac. J. Clin. Nutr., 2003; 12: 198-202
[PubMed]
[103] Torti F.M., Torti S.V.: Regulation of ferritin genes and protein. Blood, 2002; 99: 3505-3516
[PubMed] [Full Text HTML] [Full Text PDF]
[104] Trost L.B., Bergfeld W.F., Calogeras E.: The diagnosis and treatment of iron deficiency and its potential relationship to hair loss. J. Am. Acad. Dermatol., 2006; 54: 824-844
[PubMed]
[105] Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., Yoshikawa S.: Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 A. Science, 1995; 269: 1069-1074
[PubMed]
[106] Valore E.V., Ganz T.: Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol. Dis., 2008; 40: 132-138
[PubMed]
[107] Vanderpuye O.A., Kelley L.K., Smith C.H.: Transferrin receptors in the basal plasma membrane of the human placental syncytiotrophoblast. Placenta, 1986; 7: 391-403
[PubMed]
[108] Waheed A., Grubb J.H., Zhou X.Y., Tomatsu S., Fleming R.E., Costaldi M.E., Britton R.S., Bacon B.R., Sly W.S.: Regulation of transferrin-mediated iron uptake by HFE, the protein defective in hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA, 2002; 99: 3117-3122
[PubMed] [Full Text HTML] [Full Text PDF]
[109] Wąsowska-Królikowska K., Baranowski W.J.: Znaczenie żelaza w rozwoju i żywieniu niemowląt. I. Metabolizm żelaza. Medycyna Wieku Rozwojowego, 2000; 4: 65-77
[110] West A.P.Jr., Bennett M.J., Sellers V.M., Andrews N.C., Enns C.A., Bjorkman P.J.: Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J. Biol. Chem., 2000; 275: 38135-38138
[PubMed] [Full Text HTML] [Full Text PDF]
[111] Widdowson E.M., Spray C.M.: Chemical development in utero. Arch. Dis. Child., 1951; 26: 205-214
[PubMed] [Full Text PDF]
[112] Wrighting D.M., Andrews N.C.: Interleukin-6 induces hepcidin expression through STAT3. Blood, 2006; 108: 3204-3209
[PubMed] [Full Text HTML] [Full Text PDF]
[113] Yau K.W., Baylor D.A.: Cyclic GMP-activated conductance of retinal photoreceptor cells. Annu. Rev. Neurosci., 1989; 12: 289-327
[PubMed]
[114] Youdim M.B., Ben-Shachar D.: Minimal brain damage induced by early iron deficiency: modified dopaminergic neurotransmission. Isr. J. Med. Sci., 1987; 23: 19-25
[PubMed]
[115] Yu G.S., Steinkirchner T.M., Rao G.A., Larkin E.C.: Effect of prenatal iron deficiency on myelination in rat pups. Am. J. Pathol., 1986; 125: 620-624
[PubMed] [Full Text PDF]
Autorzy deklarują brak potencjalnych konfliktów interesów.