
Novel methods of cytokine detection: Real-time PCR, ELISPOT, and intracellular cytokine staining
Eliza Turlej 1Streszczenie
Cytokiny są małymi hormonopodobnymi białkami warunkującymi prawidłowe funkcjonowanie układu odpornościowego. Regulują proliferację i różnicowanie komórek, wpływają na zachodzące w organizmie procesy krwiotworzenia oraz funkcjonują jako mediatory reakcji zapalnych i odpornościowych.
Zaburzenia poziomu cytokin są widoczne w licznych chorobach np. posocznicy, zmianach jelit o charakterze zapalnym, chorobach autoimmunizacyjnych, a także w chorobie przeszczep przeciw biorcy.
Detekcję cytokin można przeprowadzić w warunkach in vivo, in vitro lub ex vivo. Istnieje wiele metod mierzących ilość wydzielonej cytokiny np. test immunoenzymatyczny ELISA oceniający sekrecję w surowicy lub w supernatantach uzyskanych po hodowli stymulowanych komórek, techniki określające poziom cytokin w komórce (barwienie immunohistochemiczne, ELISPOT, wewnątrzkomórkowe barwienie cytokin) oraz metody oceniające sekrecję na poziomie mRNA (technika RPA, hybrydyzacja in situ, Northern-blot PCR w czasie rzeczywistym).
Spośród wyżej wymienionych metod najszersze zastosowanie mają ELISPOT, metoda wewnątrzkomórkowego barwienia połączona z odczytem cytometrycznym oraz PCR w czasie rzeczywistym. Połączenie metody wewnątrzkomórkowego barwienia z analizą cytometryczną, podobnie jak i detekcja cytokinowego mRNA na skrawkach tkankowych bądź w zawiesinie komórkowej, daje możliwość oceny populacji komórek odpowiedzialnych za wydzielanie badanej cytokiny. Metody te mają duże zastosowanie kliniczne np. przy szacowaniu skuteczności szczepionek i leczenia w schorzeniach nowotworowych, a także w diagnostyce bakteriologicznej, wirusologicznej i pasożytniczej.
Słowa kluczowe:cytokina • limfocyty T • PCR w czasie rzeczywistym • ELISPOT • wewnątrzkomórkowe barwienie cytokin
Summary
Cytokines are small hormone-like proteins that play important roles in immune system control. Cytokines regulate the proliferation and differentiation of cells and hematopoiesis and act as mediators in the inflammatory reaction. Changes in cytokine levels are found in many diseases, such as sepsis, bowel inflammatory disease, autoimmune diseases, as well as graft-versus-host disease. Cytokines levels can be detected using in vivo, in vitro, and ex vivo techniques. The level of cytokine produced can be measured by immunoenzymatic test (ELISA) in supernatant after cell culture with the addition of stimulant and in plasma by techniques that measure the level of cytokine secretion in cells (e.g. immunohistochemical staining, ELISPOT, and intracellular cytokine staining), and by molecular biological methods (RPA, real-time PCR, in situ hybridization, and Northern blot). Detection of cytokine mRNA in tissues is useful in the direct determination of heterogenic populations of cytokine-producing cells. Nowadays the most frequently used methods for measuring cytokine level are ELISPOT, intracellular cytokine staining with flow cytometry detection, and real-time PCR. These methods have an important clinical role in vaccine efficacy, in viral, bacterial, and verminous diagnostics, and in determining the efficacy of cancer treatment.
Key words:cytokine • T lymphocytes • real-time PCR • ELISPOT • intracellular cytokine staining
Wykaz skrótów:
AP1 – heterodimeryczny czynnik transkrypcyjny wiążący DNA w ściśle określonym miejscu (activator protein 1); APC – komórki prezentujące antygen (antigen presenting cells); BSA – płodowa surowica cielęca (bovine serum albumin); CCCA – (cinncinati cytokine capture assay); CD – marker różnicowania (cluster of differentation); cDNA – komplementarne DNA (complementary DNA); ELISA – test immunoenzymatyczny (enzyme – linked immunosorbent assay); ELISPOT – (enzyme-linked immunosorbent spot assay); HIV – ludzki wirus niedoboru odporności (human immunodeficiency virus); HLA – ludzkie antygeny układu zgodności tkankowej (human leukocyte antigen); HRP – peroksydaza chrzanowa (horseradish peroxidase); ICS – wewnątrzkomórkowe barwienie cytokin (intracellular cytokine staining); INFγ – interferon gamma (interferon gamma); IL – interleukina (interleukin); LDA – metoda ograniczonych rozcieńczeń (limitting dilution assay); MHC – główny układ zgodności tkankowej (major histocompatibility complex); MUC1 – antygen związany z guzem, którego ekspresja widoczna jest na brzegach prawidłowego epitelium (mucin 1); mΩ – mili om; mRNA – matrycowe RNA, informacyjny kwas rybonukleinowy stanowiący matrycę do syntezy białka w procesie translacji (messenger RNA); NFAT – jądrowy czynnik aktywowanych limfocytów T, uczestniczący w aktywacji transkrypcji licznych genów, których produkty biorą udział w odpowiedzi immunologicznej (nuclear factor of activated T cell); NF-κB – czynnik jądrowy pierwotnie uważany za czynnik wymagany przy transkrypcji genów łańcucha lekkiego κ w immunoglobulinach wydzielanych przez limfocyty B (nuclear factor kappa B); NK – komórki „naturalni zabójcy“(natural killers); PBS – buforowana sól fizjologiczna (phosphonate buffer saline); PHA – fitohemaglutynina (phytohaemagglutinin); PMA – octan mirystynianu forbolu (phorbol myristate acetate); PCR – łańcuchowa reakcja polimerazy (polymerase chain reaction); PWM – mitogen szkarłatni (pokeweed mitogen); RNA – kwas rybonukleinowy (ribonuclease acid); RPA – technika ilościowej oceny mRNA (RNA-se protection assay); TCR – receptor limfocytów T (T cell receptor); TNF-α – czynnik martwicy nowotworu (tumor necrosis factor); TNFR – nadrodzina receptorów czynnika martwicy nowotworów (tumor necrosis factor-receptor).
WPROWADZENIE
Cytokiny są małymi hormonopodobnymi białkami odpowiadającymi za funkcjonowanie układu odpornościowego, oddziałującymi na neutrofile, monocyty, limfocyty oraz eozynofile, uczestniczącymi w regulacji procesów odporności i stanów zapalnych. Cytokiny biorą udział w prezentacji antygenów, wpływają na procesy różnicowania się komórek w szpiku i na ekspresję cząsteczek adhezyjnych [9,30,73].
Cechą charakterystyczną cytokin jest ich zdolność do wpływania na różne typy komórek i wywoływania w tych komórkach odmiennych działań. Cechę tę określa się mianem plejotropii. Przykładem jest interleukina 6, która oddziałuje na limfocyty B i powoduje ich przekształcenie w komórki plazmatyczne, wytwarzające immunoglobuliny, a ponadto aktywuje limfocyty T i w obecności IL-2 indukuje tworzenie limfocytów cytotoksycznych. Interleukina 6 w warunkach in vitro indukuje dojrzewanie megakariocytów oraz wzrost liczby płytek w warunkach in vivo [33,43]. Inną istotną cechą jest ich zdolność do wywoływania jednakowego efektu. Zdolność tę określa się mianem redundancji. Cytokiny mogą oddziaływać na komórki synergistycznie, co zwiększa działanie określonej interleukiny lub antagonistycznie, co osłabia działanie. Ponadto cytokiny mają zdolność do indukcji kaskady dodatnich i ujemnych sprzężeń zwrotnych.
Mogą oddziaływać na komórki autokrynnie (oddziałują wtedy na komórkę, przez którą zostały wydzielone), parakrynnie (oddziałują na sąsiednią komórkę w stosunku do komórki wydzielniczej) oraz endokrynnie (oddziałują na komórki znajdujące się w innych narządach).
Detekcja cytokin może być prowadzona w warunkach ex vivo, in vivo lub in vitro.
Badania prowadzone w warunkach in vivo pozwalają na uzyskanie aktualnych danych dotyczących obecności cytokiny w organizmie, podczas gdy badania wykonywane w warunkach ex vivo i in vitro wymagają wcześniejszej stymulacji komórek wytwarzających określoną cytokinę, co w dużej mierze ogranicza wartość uzyskanego wyniku. Często wykorzystywaną do stymulacji prowadzonej w warunkach ex vivo substancją stymulującą monocyty i makrofagi jest endotoksyna. Oprócz niej wykorzystuje się do tego celu lektyny, przeciwciała skierowane przeciwko limfocytom T oraz superantygeny [2]. Nierzadko zdarza się, że wytwarzanie cytokiny w warunkach ex vivo, w komórkach uprzednio poddanych immunizacji zwierząt, wykazuje niższy poziom od osiąganego w rzeczywistości, prowadząc do konieczności ponownej stymulacji komórek immunologicznych mitogenem albo antygenem, co wiąże się z wykorzystaniem kolejnej grupy zwierząt do przeprowadzenia eksperymentu [6].
Określenie poziomu ekspresji cytokin w żywym organizmie jest możliwe wieloma różnymi metodami np.: przez pomiar poziomu cytokiny w surowicy lub w supernatancie uzyskanym po hodowli komórek testem immunoenzymatycznym (ELISA), pomiar poziomu cytokiny w komórkach metodami histochemicznymi, cytometrycznymi i techniką ELISPOT, bądź też metodami mierzącymi poziom cytokinowego mRNA, takimi jak Northern-blot czy PCR w czasie rzeczywistym [52].
Test ELISA służy do wykrywania określonych białek w badanym materiale przy zastosowaniu przeciwciał monolub poliklonalnych skoniugowanych z enzymem. Odmianą tej techniki jest „test podwójnego wiązania” („sandwich ELISA” czyli „kanapkowa ELISA”), bardzo podobny do metody ELISPOT opisanej w następnym rozdziale. „Test podwójnego wiązania” może być wykonany metodą pośrednią lub bezpośrednią. W metodzie bezpośredniej, badany antygen wykrywa się stosując pojedyncze przeciwciało znakowane enzymem, natomiast w pośredniej wykorzystuje się pierwszo- i drugorzędowe przeciwciała, przy czym znakowane enzymem jest dopiero drugorzędowe przeciwciało, rozpoznające określony izotyp przeciwciał pierwszego rzędu. Zaletą pośredniej metody wykrywania antygenu jest możliwość wykorzystania różnych przeciwciał pierwotnych bez konieczności ich znakowania. Istnieje również możliwość wykrycia przeciwciał za pomocą określonego antygenu, jednak wyłącznie przy wykorzystaniu do tego celu testu pośredniego. Aby wykryć przeciwciała fazę stałą należy opłaszczyć antygenem, a następnie na tak przygotowane podłoże nanieść surowicę pacjenta. Po inkubacji i wypłukaniu niezwiązanych przeciwciał, dodaje się albo drugorzędowe przeciwciało albo wyznakowany ligand. Działania tego typu pozwalają nie tylko na wykrycie przeciwciała, ale nawet na określenie ich klasy, co z kolei dostarcza cennych danych epidemiologicznych.
Ograniczeniem testu ELISA jest niemożność uzyskania informacji dotyczącej liczby oraz fenotypu komórek wytwarzających badaną cytokinę. Taką możliwość daje jednak ocena sekrecji cytokiny stymulowanej lektyną lub antygenem, gdyż wytwarzana interleukina jest zatrzymywana na powierzchni komórek przez cząsteczkę dwuswoistego przeciwciała złożonego ze skoniugowanej pary monoklonalnych przeciwciał. Fizyczną izolację określonej subpopulacji komórek wydzielających cytokinę (np. komórek CD45+) umożliwia dodanie paramagnetycznych kulek skoniugowanych z przeciwciałem antycytokinowym również wyznakowanym barwnikiem fluorescencyjnym [18,49].
Badania immunohistochemiczne wykonywane na skrawkach tkankowych pozwalają na ocenę lokalizacji ekspresji cytokin w skórze, dziąsłach, jelicie grubym, nerce, mózgu oraz guzach [29]. Metody immunochemiczne polegają na wykrywaniu składników komórkowych w oparciu o reakcję antygen-przeciwciało. Zwykle wykrywa się antygen używając swoistych przeciwciał, znacznie rzadziej stosowaną odmianą reakcji jest przypadek, w którym wykrywa się w tkance przeciwciała za pomocą znakowanego antygenu. Pierwsze reakcje immunocytochemiczne prowadzono z wykorzystaniem przeciwciał znakowanych flourochromami (np. izotiocyjanianem fluoresceiny), które stopniowo zastępowano innymi znacznikami (np. enzymami – peroksydazą chrzanową czy fosfatazą alkaliczną; białkami zawierającymi w swojej strukturze cząsteczkę metalu – ferrytyną czy złotem koloidalnym). Poza tym pracowano nad wdrożeniem nowych technik, dzięki którym uzyskano zwiększenie czułości i swoistości badań. Do takich technik należą: metody pośrednie, metoda PAP (wykorzystująca nieznakowane przeciwciała), metoda ABC (wykorzystująca układ awidyna – biotyna), przeciwciała znakowane digoksygeniną oraz metoda wykorzystująca białko A połączone ze złotem koloidalnym [72]
METODA ELISPOT (ENZYME-LINKED IMMUNOSPOT ASSAY)
Pierwotnie technikę ELISPOT wykorzystywano do detekcji pojedynczych limfocytów B wytwarzających przeciwciała skierowane przeciwko swoistemu antygenowi. Później metodę tę wykorzystano do detekcji komórek wydzielających określoną cytokinę [58].
Metoda ELISPOT jest bardzo podobna do kanapkowej odmiany testu ELISA. ELISPOT stanowi połączenie techniki immunoenzymatycznej fazy stałej z krótkoterminową hodowlą komórkową trwającą od kilku godzin do kilku dni. Przebieg badania przedstawiono na ryc 1.
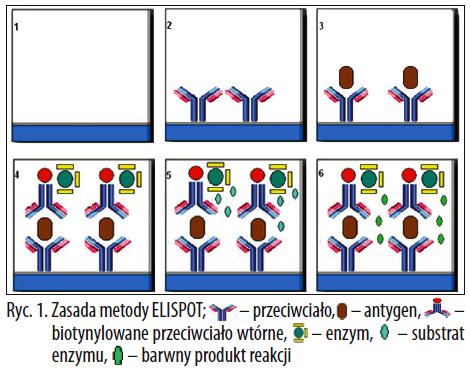
Ryc. 1. Zasada metody ELISPOT
Procedurę rozpoczyna się opłaszczeniem dołków płytki mono- lub poliklonalnym przeciwciałem swoistym dla badanej cytokiny. W następnym etapie przeciwciała umieszczone na płytce blokuje się, zazwyczaj używając do tego celu surowiczego białka, dobranego w taki sposób, aby nie wchodziło w reakcję z żadnym przeciwciałem wykorzystywanym w analizie (1,2). Do tak przygotowanej płytki dodaje się badany materiał antygenowy w taki sposób, aby uzyskać różne stężenia komórek w kolejnych dołkach płytki (3). Całość inkubuje się w temperaturze 37°C w atmosferze dwutlenku węgla w określonym czasie. Cytokina wytwarzana przez zaktywowane komórki jest wychwytywana przez przeciwciała opłaszczone na powierzchni dołków płytki (4).
Przed dodaniem poliklonalnego biotynylowanego przeciwciała swoistego dla cytokiny dołki płytki płucze się, aby usunąć pozostałości medium hodowlanego oraz niezwiązane komórki. Wtórne biotynylowane przeciwciało, wyznakowane enzymem, wiąże się z kompleksem antygen – przeciwciało pierwotne (5). Następnie dodaje się substrat dla enzymu związanego z przeciwciałem, który w wyniku reakcji przechodzi w barwny produkt (6). Najczęściej stosowanym enzymem jest peroksydaza chrzanowa, dla której substratem jest nadtlenek wodoru lub tetrametylobenzydyna, fosfataza alkaliczna (substrat: fosforan p-nitrofenolu) oraz oksydaza glukozowa (substrat: kwas 5-aminosalicylowy) [17]. W miejscu reakcji tworzą się barwne plamki ciemnego koloru. Pojedyncza plamka odpowiada jednej, wydzielającej cytokinę komórce. Plamki można zliczać metodą automatyczną z wykorzystaniem czytników płytek lub manualnie z wykorzystaniem stereomikroskopu [58].
Wynik analizy przeprowadzonej techniką ELISPOT jest uzależniony od miejscowego stężenia cytokin, jakie występuje w pobliżu komórek odpowiedzialnych za sekrecję. Jest to metoda 10-200 razy czulsza od analizy sekrecji prowadzonej metodą ELISA [56]. Wady i zalety obydwu technik przedstawia tabela 1.
Tabela 1. Podobieństwa i różnice między metodami ELISA i ELISPOT [63]

Obecnie wykorzystuje się ją do analizy limfocytów T swoistych dla antygenów związanych z komórkami czerniaka (głównie przy ocenie skuteczności opracowanych szczepionek), w badaniach nad limfocytami T swoistymi dla cytomegalowirusa u pacjentów, u których wykazano reaktywację tego wirusa, przy detekcji limfocytów B swoistych idiotypowo u osób cierpiących na szpiczaka mnogiego, a także przy oznaczaniu poziomu interferonu γ u osób chorujących na gruźlicę, u których czynnikiem stymulującym komórki do wytwarzania cytokiny są peptydy izolowane z Mycobacterium tuberculosis [3,15,35,67].
Szacowanie częstości limfocytów T swoistych dla peptydu wymaga przeprowadzenia w warunkach in vitro hodowli, w której limfocyty T są stymulowane peptydem z wykorzystaniem komórek prezentujących peptyd (APC) (tabela 2) [41].
Tabela 2. Przykłady mitogenów stosowanych do stymulacji proliferacji limfocytów T i B [61]

Metoda ELISPOT może być łączona z metodą ograniczonych rozcieńczeń (LDA – limiting dilution assay). Zaletą stosowania tego „połączenia” jest brak konieczności klonowania komórek i możliwość oceny pojedynczej komórki w populacji w oparciu o ocenę wytwarzania danej cytokiny w wybranym okresie czasu [11].
Metoda ELISPOT jest stosowana do monitorowania odpowiedzi immunologicznej u pacjentów cierpiących na nowotwory, poddawanych immunoterapii, podczas infekcji i chorób autoimmunologicznych, a także do monitorowania skuteczności szczepionek przeciwko HIV, wirusowemu zapaleniu wątroby, cholerze, półpaścowi, grypie i malarii [10,16,63].
ELISPOT jest z powodzeniem stosowany w wykrywaniu swoistej odpowiedzi immunologicznej na antygeny krętka bladego (Treponema pallidum). Jest niezastąpioną metodą w immunologii onkologicznej, diagnostyce reumatologicznej i diabetologii do oceny funkcji komórek cytotoksycznych skierowanych przeciwko wyspom trzustkowym.
Metoda ELISPOT znalazła zastosowanie również w alergologii, w diagnostyce skazy atopowej, wyprysku atopowego, alergii pokarmowej, w ocenie skuteczności immunoterapii osób uczulonych na jad owadów oraz w alergicznym zapaleniu pęcherzyków płucnych i w alergii wywołanej reakcją na leki [63].
METODA WEWNĄTRZKOMÓRKOWEGO BARWIENIA CYTOKIN
Duża grupa cytokin występuje w surowicy w ilościach niewykrywalnych metodami tradycyjnymi. Jest to wynikiem sekrecji cytokin w bardzo małych ilościach, a także ich szybkiego katabolizmu i usuwania z organizmu przez nerki. Stało się to przyczyną poszukiwania nowych metod pozwalających na dokładniejszą analizę wytwarzania cytokin i doprowadziło do odkrycia metody pierwotnie nazywanej CCCA (cincinnati cytokine capture assay). Początkowo metodę tę wykorzystywano do oceny wytwarzania IL-4 w warunkach in vivo, jednak późniejsze modyfikacje metody umożliwiły jej zastosowanie do oceny sekrecji interferonu γ. Metoda ta opiera się na obserwacji, że między analizowaną cytokiną a neutralizującym przeciwciałem monoklonalnym klasy IgG, skierowanym przeciwko badanej cytokinie tworzy się kompleks, przyczyniający się do przedłużenia okresu występowania cytokiny w surowicy. Zjawisko to ma związek z zahamowaniem ekspresji, katabolizmu i utylizacji badanej cytokiny.
Wspomniane kompleksy cytokin i monoklonalnych przeciwciał, gromadzą się w surowicy, dzięki czemu osiągają poziomy łatwo mierzalne metodami immunoenzymatycznymi. Przed wprowadzeniem do organizmu monoklonalnych przeciwciał skierowanych przeciwko badanej cytokinie, można je poddać biotynylacji ułatwiającej detekcję powstających kompleksów.
Metoda CCCA jest metodą prostą, obiektywną, czułą i swoistą w zakresie pomiaru sekrecji cytokin w warunkach in vivo, choć nie jest wolna od ograniczeń. Bardzo dużym jej ograniczeniem jest to, że monoklonalne przeciwciała przeciwcytokinowe, niekoniecznie te pożądane w toku prowadzonej analizy, mogą związać przeciwciała, hamując wyłapywanie przez nie wydzielonej cytokiny, co prowadzi do uzyskiwania wyników fałszywie negatywnych. Ponadto różne monoklonalne przeciwciała mogą oddziaływać między sobą zakłócając jednoczesny pomiar kilku cytokin, stąd zaleca się, aby metoda CCCA była wykorzystywana do pomiaru pojedynczej cytokiny u pojedynczego osobnika. Kolejnym ograniczeniem tej metody jest brak możliwości oceny precyzji oszacowania sekrecji. Obserwuje się występowanie bezpośredniego związku między ilością wydzielonej cytokiny a ilością cytokiny wykrytą w badaniu, choć niekoniecznie stosunek ten jest równy 1:1. Wzrost sekrecji cytokin powoduje spadek odsetka cytokiny związanej z przeciwciałem antycytokinowym, ponieważ albo następuje spadek wskaźnika wydzielonej cytokiny wiążącej przeciwciało przeciwcytokinowe albo wzrost związany z większym wysyceniem błony komórkowej i receptora wydzielonej cytokiny. Metoda ta jest mało przydatna do szacowania sekrecji cytokin u zwierząt większych niż myszy, ponieważ jeśli przyjmie się, że liczba znakowanych biotyną przeciwciał antycytokinowych koniecznych do wykrycia wytwarzania cytokin jest proporcjonalna do objętości płynu pozakomórkowego badanego zwierzęcia wartość poziomu cytokiny jest widocznie większa u zwierząt większych niż myszy. Niemniej jednak jej prostota i możliwość powtarzania pomiaru sekrecji cytokin w warunkach in vivo sprawia, że stanowi ona użyteczne narzędzie w badaniach nad sekrecją u małych zwierząt. Wymóg wstrzykiwania obcych przeciwciał monoklonalnych do organizmu żywego sprawia, że technika ta ma ograniczone zastosowanie u ludzi [22].
Do rozwoju oceny poziomu cytokin metodą wewnątrzkomórkowego barwienia przyczyniło się opisanie techniki pozwalającej na wykrycie cytokiny zmagazynowanej wewnątrz komórki, zastosowanie paraformaldehydu do utrwalania komórek, saponin do permabilizowania błon komórkowych oraz odkrycie pośredniej metody immunofluorescencji.
Metoda wewnątrzkomórkowego barwienia pozwala na szybką analizę dużej liczby komórek ułatwiając w ten sposób przeprowadzenie analizy statystycznej. Istotną zaletą tej techniki jest możliwość zastosowania kilku różnych fluorochromów jednocześnie, dzięki czemu można ocenić koprodukcję kilku cytokin w obrębie pojedynczej komórki [18,27,42]. Połączenie barwienia znacznikami immunofluorescencyjnymi powierzchniowych i wewnątrzkomórkowych antygenów z cytometrią przepływową stwarza możliwości analizy mieszanej populacji komórek, bez konieczności pracochłonnej separacji. Połączenie obu metod stwarza możliwość oceny stanu aktywacji komórek, ich przynależności do danej linii rozwojowej lub określonego podtypu, pozwala ocenić zdolność wiązania komórek lub tkanek, czy też migrację do miejsc zapalnych, a ponadto ocenia zdolność określonej komórki do odpowiedzi na substancję stymulującą i określa stadium różnicowania, w jakim znajduje się badana komórka [62]. Badania prowadzone z wykorzystaniem tej techniki prowadzone są na komórkach uzyskanych po izolacji wykonanej przez wirowanie w gradiencie gęstości, na krwi pełnej oraz na komórkach krioprezerwowanych. Wadą badań prowadzonych na komórkach izolowanych krwi obwodowej jest to, że wymagane są duże objętości krwi, co bardzo ogranicza wykonanie badania u dzieci. Schemat barwienia wewnątrzkomórkowego oraz jego przebieg przedstawiono na ryc. 2 i 3.
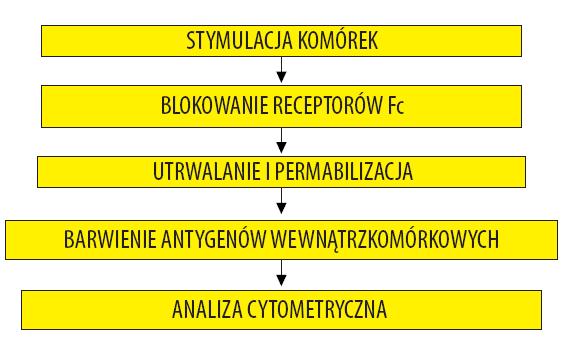
Ryc. 2. Ogólny schemat barwienia wewnątrzkomórkowego cytokin połączonego z analizą cytometryczną
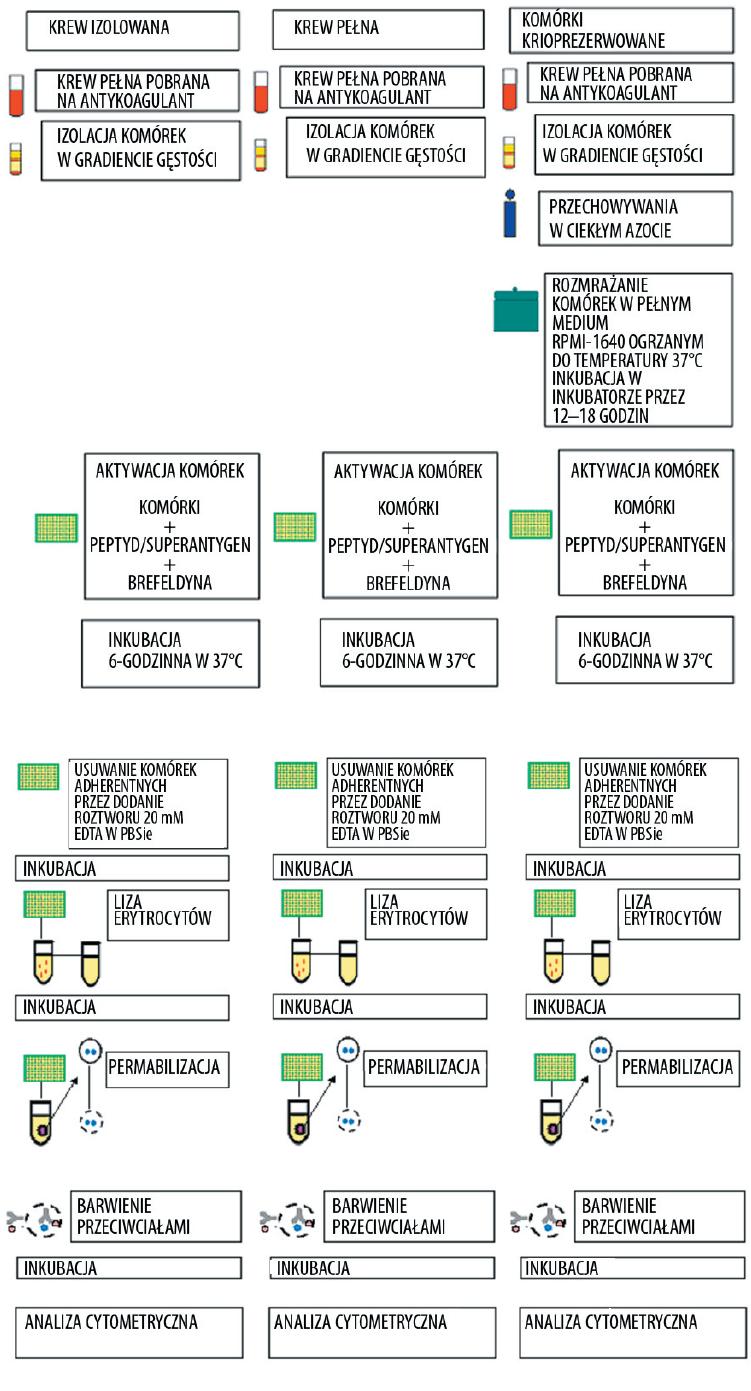
Ryc. 3. Barwienie wewnątrzkomórkowe cytokin. Porównanie schematu barwienia wewnątrzkomórkowego prowadzonego na komórkach krwi pełnej, komórkach uzyskanych po wirowaniu na gradiencie gęstości oraz na komórkach krioprezerwowanych
Analizę wytwarzania cytokin u biorców przeszczepów narządowych, których poddano długotrwałemu leczeniu immunosupresyjnemu i detekcję limfocytów T pamięci wydzielających cytokinę w odpowiedzi na swoisty antygen wirusowy przeprowadza się wykorzystując krew pełną. Technika ta nie pozwala na detekcję naiwnych limfocytów T, ponieważ ich częstość zwykle nie przekracza wartości odcięcia wynoszącej <1:10 000 komórek odpowiedzi [51,59].
Prawidłowe oszacowanie poziomu cytokiny metodą wewnątrzkomórkowego barwienia połączonego z analizą cytometryczną wymaga przeprowadzenia badań kinetycznych, pozwalających na dobranie optymalnego czasu, w którym następuje sekrecja przez komórkę efektorową. Niestymulowane limfocyty T krwi obwodowej spontanicznie wytwarzają niewielkie ilości cytokin, dlatego stymulacja komórek w warunkach in vitro jest konieczna w celu zwiększenia ekspresji genów cytokinowych [5]. Stymulację komórek prowadzi się stosując do tego celu substancje stymulujące: PMA (octan mirystynianu forbolu) łącznie z jonomycyną (jonoforem transportującym jony ołowiu w sposób selektywny) i przeciwciałami anty-CD3. Octan mirystynianu forbolu aktywuje limfocyty poprzez szlak kinazy białka C [12,20]. Jako substancję stymulującą można również wykorzystać PHA (fitohemaglutyninę), będącą lektyną wyizolowaną z czerwonej fasoli rodzaju Phaseolus vulgaris, mającą zdolności aglutynujące i mitogenne. Fitohemaglutyninę wykorzystuje się do stymulacji hodowli limfocytarnych.
Poza wymienionymi substancjami stymulującymi proliferację komórek do tego celu wykorzystuje się również superantygeny. Bakteryjne superantygeny stanowią rodzinę białek o szczególnej strukturze i sekwencji, która umożliwia pominięcie mechanizmów konwencjonalnego sposobu prezentacji antygenu. Superantygeny wiążą niezmienione białko bezpośrednio z cząsteczką MHC klasy II i z receptorem TCR w regionie zmiennym łańcucha β [37].
Przebieg procesu utrwalania i permabilizacji ma decydujący wpływ na analizę uwalniania cytokin. Najlepsze wyniki osiąga się utrwalając komórki roztworem 10% buforowanej formaliny, którą dodaje się bezpośrednio do zawiesiny komórek w buforowanej soli fizjologicznej (PBS). W kolejnym etapie przygotowane komórki zawiesza się w buforze PBS zawierającym azydek sodu oraz cielęcą surowicę płodową (BSA) i inkubuje przez noc [11]. Do utrwalania komórek można zastosować również paraformaldehyd, zapewniający dobrą permabilizację błon komórek przy jednoczesnym wzroście pośredniej fluorescencji antygenów wewnątrzkomórkowych [32].
Proces permabilizacji rozwinięto dzięki odkryciu właściwości permabilizacyjnych i hemolitycznych saponin (glikozydów pochodzenia roślinnego). Przypuszcza się, że ich właściwości wynikają z oddziaływania między saponiną a cholesterolem umiejscowionym w błonie komórkowej. W procesach permabilizacji wykonywanej dla potrzeb badań określających sekrecję cytokin po stymulacji antygenem pochodzenia wirusowego szerokie zastosowanie znalazł metanol, którego działanie polega na zwiększaniu pośredniej fluorescencji antygenów wewnątrzkomórkowych oraz na utrwalaniu struktury antygenu na długi okres czasu [4,32]. Permabilizację komórek można prowadzić również stosując Tween odpowiedzialny za utrwalanie antygenów powierzchniowych, co ma szczególne znaczenie przy selekcji limfocytów B.
W kolejnym etapie metodę wewnątrzkomórkowego barwienia cytokin przystosowano do badania komórek stymulowanych w obecności monesyny lub brefeldyny (inhibitorów transportu cytokin). Zadaniem inhibitorów transportu cytokin jest akumulacja wydzielanej cytokiny wewnątrz komórki. Monesyna jest izolowana ze Streptomyces cinnamonensis, a jej działanie polega na zaburzaniu prawidłowego gradientu jonów wodorowych H+ i sodowych Na+, umożliwiającego białkom przemieszczanie się w obrębie aparatu Golgiego, co prowadzi do akumulacji cytokiny w obrębie aparatu Golgiego i działa silnie stresogennie na komórkę. Z kolei brefeldyna A jest makrocyklicznym laktonem, syntetyzowanym z palmitynianu i wytwarzanym przez różne gatunki grzybów. Brefeldynę A wyizolowano pierwotnie z Penicillium brefeldianum, a jej działanie polega na hamowaniu wczesnej sekrecji białek między retikulum endoplazmatycznym a aparatem Golgiego. Brefeldyna A jest znacznie mniej toksyczna od monesyny, ponieważ transport pomiędzy retikulum endoplazmatycznym a aparatem Golgiego jest zachowany, z tym że wydzielane białka są zwrotnie kierowane do retikulum endoplazmatycznego [5,46,49,51].
Metoda wewnątrzkomórkowego barwienia cytokin jest wykorzystywana do detekcji odpowiedzi ze strony limfocytów T CD4+ i limfocytów T CD8+. Jest skuteczną techniką określającą cytokiny wytwarzane przez subpopulację limfocytów Th2. W przebiegu zakażeń wirusowych i analizy cytokin przez komórki stymulowane antygenami wirusowymi zauważono, że limfocyty T CD4+ odpowiadają za długoterminową kontrolę infekcji wirusowej, natomiast limfocyty T CD8+ przyspieszają terminację replikacji wirusa [19,65].
Technikę tę wykorzystuje się w pomiarach odpowiedzi na antygeny w przebiegu infekcji wirusem Epsteina-Barr, cytomegalowirusem, ludzkim wirusem niedoboru odporności, w przypadku antygenów związanych z guzem, takich jak MUC-1 (antygenu związanego z guzem, którego ekspresja jest widoczna na brzegach prawidłowego epitelium. Przeciwciała skierowane przeciwko białku MUC1 mogą blokować aktywność limfocytów cytotoksycznych w stosunku do komórek docelowych MUC1*) [26,67].
Wewnątrzkomórkowe barwienie cytokin umożliwia ocenę sekrecji cytokin w przebiegu zmian nowotworowych jajnika. Pozwala to ocenić odsetek limfocytów T CD8+ wydzielających IL-10, a ponadto umożliwia wykrycie zależności między fenotypem analizowanej komórki a wytwarzaniem IL-10. Prowadzone badania z użyciem techniki wewnątrzkomórkowego barwienia cytokin dowiodły, że limfocyty T CD8+ wydzielające tę interleukinę mają fenotyp CCR7+CD45RO+CD8+, podczas gdy limfocyty T CD8+ niewydzielające IL-10 mają fenotyp CCR7–CD45RO+CD8+. CCR7 ulega ekspresji na powierzchni limfocytów T dziewiczych (45R0) oraz pamięciowych (45RA) krążących we krwi obwodowej, aktywowanych limfocytów T zasiedlających wtórne narządy limfatyczne, a także na powierzchni dojrzałych limfocytów B i komórek dendrytycznych [66].
Innym zastosowaniem jest jakościowa ocena sekrecji cytokin przez limfocyty oraz monocyty krwi obwodowej u osób po przeszczepieniu nerek. Ekspozycja limfocytów na antygen silnie stymuluje je do sekrecji cytokin np. interferonu γ. Sygnalizacja wiązania antygenu związanego z ekspresją interferonu γ dotyczy aktywacji kinazy białka C oraz wewnątrzkomórkowego wzrostu aktywności jonów wapniowych, aktywacji aktywatora transkrypcji AP-1 (heterodimerycznego czynnika transkrypcyjnego wiążącego DNA w ściśle określonym miejscu), NF-κB (czynnika jądrowego pierwotnie uważanego za czynnik wymagany przy transkrypcji genów łańcucha lekkiego κ w immunoglobulinach wydzielanych przez limfocyty B) oraz NFAT (jądrowego czynnika aktywowanych limfocytów T biorącego udział w aktywacji transkrypcji licznych genów, których produkty biorą udział w odpowiedzi immunologicznej) [28,57,67]. Aktywacja wymienionych czynników jest niezmiernie ważna w przypadku odporności przeciwpasożytniczej, która to odporność ulega znacznemu obniżeniu przy wzroście osmolarności otoczenia. Odzwierciedleniem tego zjawiska jest osłabienie odpowiedzi immunologicznej, w przebiegu nefropatii cukrzycowej, podczas której wzrasta osmolarność pozakomórkowa, zwykle interferująca z aktywacją limfocytów. W środowisku hipertonicznym zabijanie bakterii ulega osłabieniu. Nadmierna pozakomórkowa hiperosmolarność przekraczająca 500 mΩ prowadzi do apoptozy komórek, z kolei ekspozycja na pozakomórkowy, hipertoniczny płyn o stężeniu mniejszym niż 500 mΩ ogranicza fragmentację DNA.
Wzrost osmolarności pozakomórkowej powoduje również ograniczenie wytwarzania CD69 (transmembranowej glikoproteiny zawierającej domenę wiążącą lektynę typu C), wczesnego markera aktywacji, wykazującego ekspresję na komórkach NK, limfocytach B, T, tymocytach, płytkach, eozynofilach i aktywowanych neutrofilach. Ekspresja tej cząsteczki jest regulowana na poziomie transkrypcyjnym i potranskrypcyjnym. Gen kodujący cząsteczkę CD69 jest umiejscowiony na chromosomie 12 [8,45,71]. Podobny efekt powoduje wzrost osmolarności po ekspozycji na antygen w przypadku sekrecji interferonu gamma. Mniejsze wytwarzanie interferonu γ może być wywołane inhibicją syntezy białek, spowodowaną osmotycznym obkurczeniem hepatocytów, które stymuluje ekspresję TNF-α.
DETEKCJA CYTOKIN NA POZIOMIE EKSPRESJI CYTOKINOWEGO MRNA
Detekcja cytokin na poziomie ekspresji genów opiera się na obserwacji, że obecne komórki odpornościowe wykazują słabą ekspresję cytokinowego mRNA lub nawet jej całkowity brak. Po kilku godzinach stymulacji komórek obserwuje się zauważalny wzrost poziomu mRNA wykrywany metodami biologii molekularnej. Wrażliwość klasycznych metod detekcji wystarcza do wykrycia mRNA typowych cytokin zapalnych (TNF-α, IL-6, -8) bez uprzedniej amplifikacji swoistej sekwencji kwasu nukleinowego, nie daje jednak możliwości wykrycia cytokin wydzielanych przez limfocyty T (np. IL-2 i -4). Większą czułość analizy zapewnia badanie wykonane metodą PCR [2].
Ekspresję cytokin można oszacować na poziomie mRNA z wykorzystaniem takich technik jak Northern-blot, hybrydyzacja in situ, metoda RPA (RNA-se protection assay) oraz PCR (polymerase chain reaction) i jej modyfikacji PCR w czasie rzeczywistym (real time polymerase chain reaction).
Metoda Northern-blot oraz RPA należą do metod ilościowych.
Metoda RPA (ribonuclease protection assay) jest czułą metodą wykorzystywaną do detekcji i ilościowej oceny swoistych transkryptów mRNA w mieszaninie całkowitego RNA. Przebieg badania jest bardzo prosty, zaś do wykonania badania niezbędna jest obecność rybonukleotydów, polimerazy RNA, antysensownej nici DNA i buforu zapewniającego środowisko reakcji. Próbka RNA jest syntetyzowana za pośrednictwem transkrypcji prowadzonej w warunkach in vitro. Syntetyzowana próbka jest komplementarna do sekwencji badanego genu. Próbki wykorzystywane w RPA mogą być znakowane radioaktywnym (α32P) lub biotynylowanym nukleotydem np. biotyną – N4-CTP. Radioaktywne próbki o wysokiej aktywności są przygotowywane w czasie transkrypcji, w trakcie której wszystkie nieznakowane nukleotydy są zastępowane przez nukleotydy zawierające promieniotwórczy fosfor. Znakowana próbka jest potem inkubowana z próbką całkowitego RNA lub mRNA, co ułatwia hybrydyzację komplementarnych regionów. W wyniku hybrydyzacji powstaje mieszanina zawierająca ssRNA i dsRNA, którą traktuje się rybonukleazą. Enzym ten trawi ssRNA, natomiast nie wywiera żadnego wpływu na dsRNA. W przypadku stosowania nukleotydów zawierających promieniotwórczy fosfor próbki oczyszcza się na poliakrylamidowym żelu zawierającym TBE (bufor składający się z TRIS, EDTA oraz kwasu borowego) i mocznik. Z żelu wycina się paski a kwasy nukleinowe wymywa buforem, zaś wyniki oblicza metodą scyntylacyjną.
Biotynylowane próbki są przygotowywane w podobny sposób, jednak zakłada się, że podstawieniu ulega zaledwie 60% nieznakowanych nukleotydów. Zwykle biotynylowane próbki precypitują z LiCl, a następnie są wirowane i płukane w 70% alkoholu etylowym. W przypadku tego typu próbek nie ma konieczności oczyszczania żelu. Następnie próbki zawiesza się w DEPC (diethyl pyrocarbonate – dietylowy eter kwasu pirowęglowego) i analizuje spektrofotometrem przy długości fali 260 nm [53].
W tej metodzie próbka oraz mRNA hybrydyzują w roztworze, co daje możliwość detekcji rzadkich sekwencji. Metoda RPA pozwala na wykrycie wielu transkryptów w pojedynczej próbce RNA. Metodę Northern-blot też można wykorzystać do detekcji kilku transkrytpów, jednak działanie takie wymaga przeprowadzenia przynajmniej kilku cykli reakcji. Jest to metoda około 10-krotnie mniej swoista od RPA.
Detekcja cytokinowego mRNA na skrawkach tkankowych lub w zawiesinie komórkowej pozwala na identyfikację komórek bezpośrednio odpowiedzialnych za syntezę cytokiny w heterogennej populacji komórkowej. Najlepsze wyniki osiąga się stosując jednoniciowe fragmenty RNA lub małe oligonukleotydy związane z radioizotopami lub digoksygeniną – markerem wykorzystywanym do badań kwasów nukleinowych, izolowanym z kwiatów oraz liści roślin Digitalis purpurea i Digitalis lanata, komplementarne do cytokinowego RNA [68]. Detekcja cytokinowego mRNA metodą hybrydyzacji w warunkach in situ jest metodą czułą, ale czasochłonną i drogą, a ponadto wymagającą prowadzenia ścisłej kontroli wewnętrznej.
Ocenę cytokinowego mRNA udoskonalono wprowadzając do badań nad detekcją metodę PCR i jej modyfikację PCR w czasie rzeczywistym [6]. Reakcja PCR umożliwia powielanie matrycowej cząsteczki. Do jej prawidłowego przebiegu konieczna jest obecność: matrycy, enzymu przeprowadzającego reakcję (termostabilnej polimerazy Taq, izolowanej z termofilnych bakterii Thermus aquaticus), oligonukleotydowych starterów, nukleotydów, odpowiedniego buforu zawierającego jony magnezu.
Reakcja PCR to powtarzające się cykle, z których każdy składa się z następujących po sobie etapów: denaturacji dwuniciowego DNA, przyłączania starterów oraz syntezy nowych, komplementarnych nici. Detekcja zamplifikowanego DNA polega na elektroforezie kwasów nukleinowych w obecności bromku etydyny i następującej analizie prążków, widocznych po poddaniu żelu działaniu światła ultrafioletowego.
Technika PCR w czasie rzeczywistym umożliwia badanie poziomu ekspresji genu w sposób względny lub bezwzględny. Bezwzględne pomiary ilościowe wymagają sporządzenia krzywej wzorcowej, którą uzyskuje się przez amplifikację próbek o znanej liczbie kopii badanego genu. Pomiary takie zastosowano w badaniach poziomu ekspresji białka prionowego w mózgowiu owiec cierpiących na trzęsawkę.
Względne badanie poziomu ekspresji genu polega na porównaniu poziomu ekspresji badanego genu z poziomem genu referencyjnego (tzw. kontrolą wewnętrzną), przy czym zakłada się, że poziom genu referencyjnego ma stałą wartość i pozostaje niezmieniony w trakcie całej analizy [48,50] Rolę kontroli wewnętrznej pełnią zwykle geny cyklofiliny, podjednostki 18S rRNA, receptora transferynowego czy też β-aktyny. Tego rodzaju badania zastosowano m.in. w celu określenia ekspresji czynników wzrostu neuroleukiny oraz izoform homologa fibroblastycznego czynnika wzrostu w komórkach nerwowych myszy zainfekowanych wirusem wścieklizny [48].
W metodzie PCR w czasie rzeczywistym wykorzystuje się barwniki emitujące światło o określonej długości w wyniku związania się z dwuniciową cząsteczką DNA. Przykładem takiego barwnika jest SYBR Green I – cyjaninowy barwnik fluorescencyjny, stosowany do wybarwiania jąder komórkowych oraz chromosomów w badaniach prowadzonych w mikroskopie fluorescencyjnym. Wadą zastosowania tego barwnika jest jego nieswoiste wiązanie się ze strukturą powstającą w wyniku połączenia ze sobą starterów wykazujących komplementarność względem siebie. Rozwiązaniem tego problemu jest przeprowadzenie badań zmierzających do ustalenia wartości temperatury topnienia [21,69].
Istotnym elementem reakcji PCR jest odpowiedni dobór sond. Pierwszymi sondami, które wykorzystano w metodzie PCR w czasie rzeczywistym były krótkie oligonukleotydy zawierające na jednym końcu cząsteczkę reporterową zaś na drugim końcu cząsteczkę wygaszającą emisję fluorescencji. W przebiegu reakcji sonda wiązała się z sekwencją komplementarną a następnie była degradowana w wyniku działania polimerazy Taq, dzięki czemu fluorochrom ulegał odseparowaniu od cząsteczki wygaszającej i emitował fluorescencję [36,64]. Obserwacja oraz pomiar fluorescencji wiążą się ze zjawiskiem FRET (fluorescence resonance energy transfer) polegającym na przenoszeniu energii między dwoma chromoforami w sposób odmienny od promieniowania. Wzbudzony donor przekazuje energię wzbudzenia akceptorowi, który znajduje się w odległości nieprzekraczającej 10 nm. Jeśli donor i akceptor różnią się widmem absorpcji i emisji promieniowania, a są umiejscowione w odległości nie większej niż 10 nm to wzbudzenie donora wiązką o długości fali odpowiadającej maksimum jego absorpcji daje emisję fali, której długość jest równa maksimum fali emitowanej przez akceptor. Jeśli natomiast odległość między donorem a akceptorem przekracza 10 nm, obserwuje się tylko emisję fali przez cząsteczkę donora z charakterystycznym dla niego maksimum absorpcji [50].
Do niewątpliwych zalet PCR w czasie rzeczywistym należy prostota wykonania oraz to, że do badania wystarczają zaledwie niewielkie ilości mRNA, dzięki czemu jest stosowana z dużym powodzeniem do analizy bioptatów lub lizatów pobranych w trakcie mikrodyssekcji [11]. Przedstawienie końcowego wyniku analizy nie wymaga dodatkowych działań, dzięki czemu zapobiega się niepotrzebnemu zanieczyszczeniu próbek. Wadą metody jest podatność reakcji PCR na inhibicję przez związki zawarte w próbkach biologicznych. Przykładem związków negatywnie oddziałujących na reakcję PCR jest mocznik i hemoglobina. Błędne wyniki są uzyskiwane często w następstwie ludzkich pomyłek związanych z niewłaściwym wykorzystaniem metody lub nieprawidłową oceną otrzymanych wyników.
Technikę PCR w czasie rzeczywistym stosuje się w praktyce klinicznej z dużym powodzeniem np.: przy szacowaniu ekspresji cytokinowego mRNA w węzłach chłonnych oraz w komórkach osób cierpiących na chłoniaka Hodgkina [40]. Metoda umożliwia detekcję zmian wywołanych uszkodzeniem prawidłowego genu. Ponadto metoda ta znalazła zastosowanie w diagnostyce molekularnej przy wykrywaniu licznych patogenów: grzybów, wirusów i bakterii w tkankach [12]. W przypadku infekcji wirusowych pozwala oszacować efektywność prowadzonej terapii przeciwwirusowej oraz postęp choroby. W odniesieniu do infekcji bakteryjnych pozwala na wykrycie i identyfikację bakterii, dzięki czemu daje możliwość dobrania skutecznej, celowanej antybiotykoterapii. Najczęściej reakcję PCR w czasie rzeczywistym stosuje się w badaniach prowadzonych nad drobnoustrojami, takimi jak: Mycobacterium tuberculosis, Legionella pneumophila, Listeria monocytogenes i Neisseria gonorhoeae. Natomiast analiza mutacji pozwala monitorować oporność lekową wśród Staphylococcus epidermis, Helicobacter pylori, Enterococcus faecalis i Enterococcus faecium [64].
Zdolność reakcji PCR w czasie rzeczywistym do identyfikacji sekwencji DNA ma bardzo duże znaczenie w onkologii klinicznej, ponieważ dzięki tej reakcji możliwe jest wykrycie, a czasem nawet ilościowe oszacowanie translokacji w chromosomach lub fuzji ich transkryptów genów głównie u pacjentów z minimalną chorobą resztkową (np. pomiar produktów fuzji genów AML-1/MTG 8 u osób cierpiących na ostrą białaczkę szpikową, rearanżacje kilku produktów genów w ostrej białaczce limfoblastycznej, czy też u pacjentów odpowiadających na leczenie INF-α przez pomiar produktów fuzji genów BCR-ABL u osób z przewlekłą białaczką szpikową).
Reakcja PCR w czasie rzeczywistym może być również wykorzystywana do określania kinetyki ekspresji genów docelowych w odpowiedzi na enzymy, warunkujące dystrybucję i funkcjonowanie leku [64].
Głównym czynnikiem ograniczającym reakcję jest konieczność zastosowania polimerazy. Problem ten można jednak rozwiązać stosując odwrotną transkryptazę ułatwiającą przepisanie RNA na cDNA. Metoda ta nosi nazwę RT real-time PCR (reverse transcriptase real-time polymerase chain reaction), a jej zastosowanie pozwala na wykrywanie w badanym materiale wirusów RNA [12].
PODSUMOWANIE
Cytokiny są białkami o strukturze zbliżonej do hormonów. Sekrecja cytokin oraz ich oddziaływanie na organizm warunkuje jego prawidłowe funkcjonowanie. Różnorodność funkcji pełnionych przez cytokiny sprawia, że szacowanie poziomu cytokin w organizmie nabiera znaczenia klinicznego.
Zaburzenia poziomu cytokin są widoczne w różnych chorobach np. w przebiegu posocznicy, chorób jelit o podłożu zapalnym, chorób autoimmunizacyjnych, w przebiegu choroby Crohna, reumatoidalnym zapaleniu stawów czy też w chorobie przeszczep przeciw biorcy, stanowiącej częste powikłanie przeszczepów szpiku i komórek krwiotwórczych szpiku oraz komórek hematopoetycznych krwi pępowinowej. Niski poziom wytwarzanych cytokin w komórkach krwi pępowinowej wiąże się z mniejszym ryzykiem wystąpienia choroby przeszczep przeciw biorcy. Dzięki ocenie poziomu cytokin można monitorować progresję choroby [7,62].
Zaburzenia poziomu cytokin mają związek z zaburzeniami hematopoezy, procesu odpowiedzialnego za powstawanie dojrzałych krwinek, a ponadto wiążą się z nieprawidłowym rozwojem narządów chłonnych, z selektywnie przebiegającą aktywacją leukocytów, a także z występowaniem stanów zapalnych, chorobami alergicznymi, procesem gojenia się ran, angiogenezą naczyń krwionośnych oraz z powstawaniem przerzutów nowotworowych [25,44].
Spośród różnych metod mających na celu detekcję zmian poziomu cytokin w organizmie najszersze zastosowanie ma technika ELISPOT, metoda wewnątrzkomórkowego barwienia cytokin oraz detekcja cytokinowego mRNA oparta na reakcji PCR w czasie rzeczywistym.
Wykorzystanie metody wewnątrzkomórkowego barwienia cytokin połączonego z odczytem cytometrycznym okazało się pomocne w badaniach nad rolą poszczególnych cytokin w różnych stanach chorobowych włączając mononukleozę, zaburzenia odporności, syndrom hiper-IgE, stwardnienie rozsiane oraz infekcję wirusem HIV, a także w odniesieniu do schorzeń pasożytniczych np. leiszmaniozy [39,54]. Metoda ta jest oprócz techniki ELISPOT jedną z dwóch najczęściej wykorzystywanych metod do oceny immunogenności szczepionek w doświadczeniach, w których ocenia się poziom sekrecji cytokin przez limfocyty T [38]. Zaletą każdej z wymienionych metod jest różnorodność zastosowań, co w znacznym stopniu determinuje ich rozwój. W przypadku techniki wewnątrzkomórkowego barwienia cytokin prowadzi się prace nad konstrukcją cytometrów przepływowych pozwalających na jednoczesną analizę coraz większej liczby parametrów w tym samym czasie. W odniesieniu do metody PCR w czasie rzeczywistym pracuje się nad udoskonaleniem sond oraz starterów, nowymi kombinacjami fluorochromów. Obiecujące rezultaty daje zastosowanie cząsteczek NFQ (non-fluorescent quencher) wygaszających energię a przy tym niewywołujących emisji fluorescencji.
PIŚMIENNICTWO
[1] Andreasen O.S, Christensen J.E., Marker M., Thomsen A.R.: Role of CD40 ligand and CD28 in induction and maintenance of antiviral CD8+ effector T cell responses. J. Immunol., 2000; 164: 3689-3697
[PubMed] [Full Text HTML] [Full Text PDF]
[2] Asadullah K., Sterry W., Volk H.D.: Analysis of cytokine expression in dermatology. Arch. Dermatol., 2002; 138: 1189-1196
[PubMed] [Full Text HTML] [Full Text PDF]
[3] Asai T., Storkus W.J., Whiteside T.L.: Evaluation of the modified ELISPOT assay for gamma interferon production in cancer patients receiving antitumor vaccines. Clin. Diagn. Lab. Immunol., 2000; 7: 145-154
[PubMed] [Full Text HTML] [Full Text PDF]
[4] Bachran C., Sutherland M., Heisler I., Hebestreit P., Melzig M., Fuchs H.: The saponin-mediated enhanced uptake of targeted saporin-based drugs is strongly dependent on the saponin structure. Exp. Biol. Med., 2006; 231: 412-420
[PubMed] [Full Text HTML] [Full Text PDF]
[5] Baran J., Kowalczyk D., Ozóg M., Zembala M.: Three-color flow cytometry detection of intracellular cytokines in peripheral blood mononuclear cells: comparative analysis of phorbol myristate acetate-ionomycin and phytohemagglutinin stimulation. Clin. Diagn. Lab. Immunol., 2001; 8: 303-313
[PubMed] [Full Text HTML] [Full Text PDF]
[6] Bienvenu J.A., Monneret G., Gutowski M.C., Fabien N.: Cytokine assays in human sera and tissues. Toxicology, 1998; 129: 55-61
[PubMed]
[7] Bogunia-Kubik K.: Wytwarzanie cytokin przez komórki krwi obwodowej i pępowinowej – porównanie i próba wyjaśnienia istniejących różnic. Post. Hig. Med. Dośw., 2001; 55: 629-641
[PubMed]
[8] Bogunia-Kubik K., Natarajan P., Madrigal J.A., Cohen S.B.: The effect of cord blood sera on CD69 expression. Immunol. Lett., 2002; 84: 77-80
[PubMed]
[9] Borish L.C., Steinke J.W.: 2. Cytokines and chemokines. J. Allergy Clin. Immunol., 2003; 111: S460-S475
[PubMed]
[10] Boulet S., Ndongala M.L., Peretz Y., Boisvert M.P., Boulassel M.R., Tremblay C., Routy J.P., Sekaly R.P., Bernard N.F.: A dual color ELISPOT method for the simultaneous detection of IL-2 amd INF-gamma HIV-specific immune responses. J. Immunol. Methods, 2007; 320: 18-29
[PubMed]
[11] Budhia S., Haring L.F., McConnell I., Blacklaws B.A.: Quantitation of ovine cytokine mRNA by real-time RT-PCR. J. Immunol. Methods, 2006; 309: 160-172
[PubMed]
[12] Bustin S., Benes V., Nolan T., Pfaffl M.: Quantitative real-time RT-PCR – a perspective. J. Mol. Endocrinol., 2005; 34: 597-601
[PubMed] [Full Text HTML] [Full Text PDF]
[13] Carter L.L., Swain S.L.: Single cell analyses of cytokine production. Curr. Opin. Immunol., 1997; 9: 177-182
[PubMed]
[14] Christensen J.E., Christensen J.P., Kristensen N.N., Hansen N.J., Stryhn A., Thomsen A.R.: Role of CD28 co-stimulation in generationand maintenance of virus-specific T cells. Int. Immunol., 2002; 14: 701-711
[PubMed] [Full Text HTML] [Full Text PDF]
[15] Comin-Anduix B.,Gualberto A., Glaspy J.A., Seja E., Ontiveros M., Reardon D.L., Renteria R., Englahner B., Economou J.S., Gomez-Navarro J., Ribas A.: Definition of an immunologic response using the major histocompatibility complex tetramer and enzyme-linked immunospot assays. Clin. Cancer. Res., 2006; 12: 107-116
[PubMed] [Full Text HTML] [Full Text PDF]
[16] Cox J.H., Ferrari G., Janetzki S.: Measurement of cytokine release at the single cell level using the ELISPOT assay. Methods, 2006; 38: 274-282
[PubMed]
[17] Czerkinsky C., Nilsson L., Nygren H., Ouchterlony O., Tarkowski A.: A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells. J. Immunol. Methods, 1983; 65: 109-121
[PubMed]
[18] Desombere I., Meuleman P., Rigole H., Willems A., Irsch J., Leroux-Roels G.: The interferon gamma secretion assay: a reliable tool to study interferon gamma production at the single cell level. J. Immunol. Methods, 2004; 286: 167-185
[PubMed]
[19] Egli A., Binet I., Binggeli S., Jager C., Dumoulin A., Schaub S., Steiger J., Sester U., Sester M., Hirsch H.H.: Cytomegalovirus-specific T-cell responses and viral replication in kidney transplant recipients. J. Transl. Med., 2008; 6: 29
[PubMed] [Full Text HTML] [Full Text PDF]
[20] Erdahl W.L., Chapman C.J., Taylor R.W., Pfeiffer D.R.: Ionomycin, a carboxylic acid ionophore, transports Pb2+ with high selectivity. J. Biol. Chem., 2000; 275: 7071-7079
[PubMed] [Full Text HTML] [Full Text PDF]
[21] Espy M.J., Uhl J.R., Sloan L.M., Buckwalter S.P., Jones M.F., Vetter E.A., Yao J.D., Wengenack N.L., Rosenblatt J.E., Cockerill F.R.3rd, Smith T.F.: Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin. Mirobiol. Rev., 2006; 19: 165-256
[PubMed] [Full Text HTML] [Full Text PDF]
[22] Finkelman F.D., Morris S.C.: Development of an assay to measure in vivo cytokine production in the mouse. Int. Immunol., 1999; 11: 1811-1818
[PubMed] [Full Text HTML] [Full Text PDF]
[23] Gajewski T.: Monitoring specific T-cell responses to melanoma vaccines: ELISPOT, tetramers, and beyond.. Clin. Diagn. Lab. Immunol., 2000; 7: 141-144
[PubMed] [Full Text HTML] [Full Text PDF]
[24] Gennery A.R., Khawaja K., Veys P., Bredius R.G., Notarangelo L.D., Mazzolari E., Fischer A., Landais P., Cavazzana-Calvo M., Fredrich W., Fasth A., Wulffraat N.M., Matthes-Martin S., Bensoussan D., Bordigoni P., Lange A., Pagliuca A., Andolina M., Cant A.J., Davies E.G.: Treatment of CD40 ligand deficiency by hematopoietic stem cell transplantation: A survey of the European experience, 1993-2002. Blood, 2004; 103: 1152-1157
[PubMed]
[25] Gieryng A., Bogunia-Kubik K.: Znaczenie interakcji pomiędzy SDF-1 i jego receptorem w hematopoezie i mobilizacji macierzystych komórek hematopoetycznych do krwi obwodowej. Post. Hig. Med. Dośw., 2007; 61: 369-383
[PubMed] [Full Text PDF] [Full Text PDF]
[26] Gong J., Chen D., Kashiwaba M., Li Y., Chen L., Takeuchi H., Qu H., Rowse G.J., Gendler S.J., Kufe D.: Reversal of tolerance to human MUC1 antigen in MUC1 transgenic mice immunized with fusions of dendritic and carcinoma cells. Proc. Natl. Acad. Sci. USA, 1998; 95: 6279-6283
[PubMed] [Full Text HTML] [Full Text PDF]
[27] Hebart H., Daginik S., Stevanovic S., Grigoleit U., Dobler A., Baur M., Rauser G., Sinzer C., Jahn G., Loeffler J., Kanz L., Rammensee H.G., Einsele H.: Sensitive detection of human cytomegalovirus peptide-specific cytotoxic T-lymphocyte responses by interferon-γ-enzyme-linked immunospot assay and flow cytometry in healthy individuals and in patients after allogeneic stem cell transplantation. Blood, 2002; 99: 3830-3837
[PubMed]
[28] Hogan P., Chen L., Nardone J., Rao A.: Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev., 2003; 17: 2205-2232
[PubMed] [Full Text HTML] [Full Text PDF]
[29] Hopkins S.J.: The pathophysiological role of cytokines. Leg. Med. (Tokyo), 2003; 5: S45-S57
[PubMed]
[30] Horohov D.W.: Equine cytokines: past, present and future. Equine J. Vet. Science, 2003; 23: 331-332
[31] House R.V.: Cytokines measurement techniques for assessing hypersensitivity. Toxicology, 2001; 158: 51-58
[PubMed]
[32] Imbert-Marcille B.M., Coste-Burel M., Robillard N., Foucaud-Gamen J., Billaudel S., Drouet E.: Sequential use of paraformaldehyde and methanol as optimal conditions for the direct quantification of ZEBRA and Rta antigens by flow cytometry. Clin. Diagn. Lab. Immunol., 2000; 7: 206-211
[PubMed] [Full Text HTML] [Full Text PDF]
[33] Karabon L., Wysoczańska B., Bogunia-Kubik K., Suchnicki K., Lange A.: IL-6 and IL-10 promoter gene polymorphisms of patients and donors of allogeneic sibling hematopoietic stem cell transplants associate with the risk of acute graft-versus-host-disease. Hum. Immunol., 2005; 66: 700-710
[PubMed]
[34] Lange A.: Transplantologia. Kowalski M.: Immunologia kliniczna. Oficyna Wydawnicza Mediton, Łódź 2000
[35] Lawn S., Bangani N., Vogt M., Bekker L., Badri M., Ntobongwana M., Dockrell H., Wilkinson R., Wood R.: Utility of interferon-γ ELISPOT assay responses in highly tuberculosis-exposed patients with advanced HIV infection in South Africa. BMC Infect. Dis., 2007; 7: 99
[PubMed] [Full Text HTML] [Full Text PDF]
[36] Listvanova S., Temmerman S., Stordeur P., Verscheure V., Place S., Zhou L., Locht C., Mascart F.: Optimal kinetics for quantification of antigen-induced cytokines in human peripheral blood mononuclear cells by real-time PCR and by ELISA. J. Immunol. Methods, 2003; 281: 27-35
[PubMed]
[37] Llewelyn M., Cohen J.: Superantigens: microbial agents that corrupt immunity. Lancet Infect. Dis., 2002; 2: 156-162
[PubMed]
[38] Maecker H.T., Rinfret A., D’Souza P., Darden J., Roig E., Landry C., Hayes P., Birungi J., Anzala O., Garcia M., Harari A., Frank I., Baydo R., Baker M., Holbrook J., Ottinger J., Lamoreaux L., Epling C.L., Sinclair E., Suni M.A., Punt K., Calarota S., El-Bahi S., Alter G., Maila H., Kuta E., Cox J., Gray C., Altfeld M., Nougarede N., Boyer J., Tussey L., Tobery T., Bredt B., Roederer M., Koup R., Maino V.C., Weinhold K., Pantaleo G., Gilmour J., Horton H., Sekaly R.P.: Standardization of cytokine flow cytometry assays. BMC Immunol., 2005; 6: 13
[PubMed] [Full Text HTML] [Full Text PDF]
[39] Magee C.C., Denton M.D., Womer K.L., Khoury S.J., Sayegh M.H.: Assessment by flow cytometry of intracellular cytokine production in the peripheral blood cells of renal transplant recipients. Clin. Transplant., 2004; 18: 395-401
[PubMed]
[40] Malec M., Soderqvist M., Sirsjo A., MacNamara B., Lewin N., Sjoberg J., Bjórkholm M., Porwit-MacDonald A.: Real-time polymerase chain reaction determination of cytokine mRNA expression profiles in Hodgkin’s lymphoma. Haematologica, 2004; 89: 679-685
[PubMed] [Full Text PDF]
[41] Markovic S.N., Newala W.K., Uhl C.B., Celis E., McKean D.: A reproducible method for the enumeration of functional (cytokine producing) versus non-functional peptide-specific cytotoxic T lymphocytes in human peripheral blood. Clin. Exp. Immunol., 2006; 145: 438-447
[PubMed] [Full Text HTML] [Full Text PDF]
[42] Mascher B., Schlenke P., Seyfarth M.: Expression and kinetics of cytokines determined by intracellular staining using flow cytometry. J. Immunol. Methods, 1999; 223: 115-121
[PubMed]
[43] Mazur G., Bogunia-Kubik K., Wróbel T., Karabon L., Polak M., Kuliczkowski K., Lange A.: IL-6 and IL-10 promoter gene polymorphisms do not associate with the susceptibility for multiple myeloma. Immunol. Lett., 2005; 96: 241-246
[PubMed]
[44] Mazur G., Jaskuła E., Kryczek I.: Udział chemokin w chorobach nowotworowych. Adv. Clin. Ex. Med., 2004; 13: 315-325
[45] Murata K., Inami M., Hasegawa A., Kubo S., Kimura M., Yamashita M., Hosokawa H., Nagao T., Suzuki K., Hashimoto K., Shinkai H., Koseki H., Taniguchi M., Ziegler S.F., Nakayama T.: CD69-null mice protected from arthiritis induced with anti-type II collagen antibodies. Int. Immunol., 2003; 15: 987-992
[PubMed] [Full Text HTML] [Full Text PDF]
[46] Nylander S., Kalies I.: Brefeldin A, but not monesin, completely blocks CD69 expression on mouse lymphocytes: efficacy of inhibitors of protein secretion in protocols for intracellular cytokine staining by flow cytometry. J. Immunol. Methods, 1999; 224: 69-76
[PubMed]
[47] O’Neil Andersen N.J., Lawrence D.A.: Differential modulation of surface and intracellular protein expression by T cells after stimulation in the presence of monesin or brefeldin A. Clin. Diagn. Lab. Immunol., 2002; 9: 243-250
[PubMed] [Full Text HTML] [Full Text PDF]
[48] Orłowska A., Smreczak M., Trębas P., Żmudziński J.: Zastosowanie real time PCR z uwzględnieniem przydatności w diagnostyce wścieklizny. Medycyna Wet., 2008; 64: 1280-1282
[Full Text PDF]
[49] Pittet M.J., Zippelius A., Speiser D.E., Assenmacher M., Guillaume P., Valmori D., Lienard D., Lejeune F., Cerottini J.C., Romero P.: Ex vivo INF-γ secretion by circulating CD8 T lymphocytes: Implications of a novel approach for T cell monitoring in infectious and malignant diseases. J. Immunol., 2001; 166: 7634-7640
[PubMed] [Full Text HTML] [Full Text PDF]
[50] Radwan M., Jonszta D., Vnenchak-Kosz M.: Metoda PCR w czasie rzeczywistym (real-time PCR) – wyzwania i perspektywy. Diagnosta Laboratoryjny, 2008; 2: 10-17
[51] Reinsmoen N.L.: Cellular methods used to evaluate the immune response in transplantation. Tissue Antigens, 2002; 59: 241-250
[PubMed]
[52] Rodriguez L., Gonzales C., Flores L., Jimenez-Zamudio L., Graniel J., Ortiz R.: Assessment by flow cytometry of cytokine production in malnourished children. Clin. Diagn. Lab. Immunol., 2005; 12: 502-507
[PubMed] [Full Text HTML] [Full Text PDF]
[53] Rottman J.B.: The ribonuclease protection assay: a powerful tool for the veterinary pathologist. Vet. Pathol., 2002; 39: 2-9
[PubMed] [Full Text HTML] [Full Text PDF]
[54] Santiago M.A., De Luca P.M., Bertho A.L., Azeredo-Coutinho R.B., Coutinho S.G.: Detection of intracytoplasmic cytokines by flow cytometry. Mem. Inst. Oswaldo Cruz, Rio de Janeiro, 2000; 95: 401-402
[PubMed] [Full Text HTML] [Full Text PDF]
[55] Schacklett BL.: Beyond 51Cr release: New methods for assessing HIV-1-specific CD8+ T cell responses in peripheral blood and mucosal tissues. Clin. Exp. Immunol., 2002; 130: 172-182
[PubMed] [Full Text HTML] [Full Text PDF]
[56] Scheibenbogen C., Lee K.H., Stevanovic S., Witzens M., Willhauck M., Waldmann V., Naeher H., Rammensee H.G., Keilholz U.: Analysis of the T cell response to tumor and viral peptide antigens by an INFγ-ELISPOT assay. Int. J. Cancer, 1997; 71: 932-936
[PubMed] [Full Text PDF]
[57] Schiff R., Reddy P., Ahotupa M., Coronado-Heinsohn E., Grim M., Hilsenbeck S., Lawrence R., Deneke S., Herrera R., Chamness G.S., Fuqua S.A., Brown P.H., Osborne C.K.: Oxidative stress and AP-1 activity in tamoxifen resistant breast tumors in vivo. J. Natl. Cancer Inst., 2000; 92: 1926-1934
[PubMed] [Full Text HTML] [Full Text PDF]
[58] Schmittel A., Keilholz U., Scheinbenbogen C.: Evaluation of the interferon-γ ELISPOT – assay for the quantification of peptide specific T lymphocytes from peripheral blood. J. Immunol Methods, 1997; 210: 167-174
[PubMed]
[59] Sewell W.A., North M.E., Webster A.D., Farrant J.: Determination of intracellular cytokines by flow-cytometry following whole-blood culture. J. Immunol. Methods, 1997; 209: 67-74
[PubMed]
[60] Smit W.M., Rijnbeek M., Van Bergen C.A., Willemze R., Falkenburg J.H.: Generation of leukemia-reactive cytotoxic T lymphocytes from HLA-identical donors of patients with chronic myeloid leukemia using modifications of a limiting dilution assay. Bone Marrow Transplant., 1998; 21: 553-560
[PubMed] [Full Text HTML] [Full Text PDF]
[61] Stokłosa S.: Hodowla komórek i tkanek. Wydawnictwo Naukowe PWN, Warszawa 2006
[62] Sullivan K.E., Cuttili J., Piliero L.M., Ghavimi-Alagha D., Starr S.E., Campbell D.E., Douglas S.D.: Measurement of cytokine secretion, intracellular protein expression, and mRNA in resting and stimulated peripheral blood mononuclear cells. Clin. Diagn. Lab. Immunol., 2000; 7: 920-924
[PubMed] [Full Text HTML] [Full Text PDF]
[63] Śpiewak R.: Test immunoenzymatyczny ELISPOT. Perspektywy zastosowań w alergologii i immunologii. Immunologia, 2007; 4: 77-81
[64] Valasek M.A., Repa J.J.: The power of real-time PCR. Adv. Physiol. Educ., 2005; 29: 151-159
[PubMed] [Full Text HTML] [Full Text PDF]
[65] Volk H.D., Kern F.: Insights into the specificity and function of (allo)antigen-reactive T cells. Am. J. Transplant., 2001; 1: 109-114
[PubMed]
[66] Wei S., Kryczek I., Zou L., Daniel B., Cheng P., Mottram P., Curiel T., Lange A., Zou W.: Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res., 2005; 65: 5020-5026
[PubMed] [Full Text HTML] [Full Text PDF]
[67] Whiteside T.L., Zhao Y., Tsukishiro T., Elder E.M., Gooding W., Baar J.: Enzyme-linked immunospot, cytokine flow cytometry, and tetramers in the detection of T-cell responses to a dendritic cell-based multipeptide vaccine in patients with melanoma. Clin. Cancer Res., 2003; 9: 641-649
[PubMed] [Full Text HTML] [Full Text PDF]
[68] Wyroba J., Wiejak J., Cywińska A., Surmach L.: Digoksygenina- nieradioaktywny marker w badaniach kwasów nukleinowych. Post. Biol. Komórki, 1998; 25: 125-133
[69] Yin J.L., Shackel N.A., Zekry A., Mcguinness P.H., Richards C., Putten K.V., Mccaughan G.W., Eris J.M., Bishop G.A.: Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) for measurement of cytokine and growth factor mRNA expression with fluorogenic probes or SYBR Green I. Immunol. Cell. Biol., 2001; 79: 213-221
[PubMed]
[70] Young N., Roelen D., Dallman M., Wood K., Morris P., Welsh K.: Enumeration of human alloreactive helper T lymphocyte precursor frequencies by limiting dilution analysis of interleukin-2 production. J. Immunol. Methods, 1996; 195: 33-41
[PubMed]
[71] Yu X., Matsui T., Otsuka M., Sekine T., Yamamoto K., Nishioka K., Kato T.: Anti-CD69 autoantibodies cross-react with low density lipoprotein receptor-related protein 2 in systemic autoimmune diseases. J. Immunol., 2001; 166: 1360-1369
[PubMed] [Full Text HTML] [Full Text PDF]
[72] Zabel M.: Immunocytochemia, PWN Warszawa 1999
[73] Zlotnik A., Yoshie O.: Chemokines: A new classification system and their role in immunity. Immunity, 2000; 12: 121-127
[PubMed] [Full Text HTML] [Full Text PDF]
Autorka deklaruje brak potencjalnych konfliktów interesów.