
Role of tumor microenvironment in the formation and progression of skin melanoma
Magdalena Olbryt 1Streszczenie
Czerniak jest nowotworem rozwijającym się z komórek barwnikowych umiejscowionych w skórze, w rzadszych przypadkach w oku oraz błonach śluzowych. Należy do grupy najbardziej agresywnych nowotworów, opornych na standardowe leczenie onkologiczne. Rozwija się głównie pod wpływem promieniowania UV. Około 10% przypadków ma podłoże dziedziczne. Główną przyczyną transformacji nowotworowej melanocytów są mutacje w genach kodujących białka regulujące proliferację, wzrost, cykl komórkowy oraz śmierć komórki. Jakkolwiek utrwalone zmiany w DNA melanocytów są podstawą powstawania czerniaka, coraz lepiej poznawana jest rola tzw. mikrośrodowiska skóry.
Prawidłowe komórki skóry, czyli keratynocyty, fibroblasty, komórki śródbłonkowe oraz komórki układu odpornościowego wraz z melanocytami tworzą mikrośrodowisko. Jego zachwiana homeostaza może sprzyjać rozwojowi czerniaka. Proces transformacji jest zapoczątkowany przez zaburzenie kontaktu keratynocytów i melanocytów. W dalszym etapie miofibroblasty stymulują proliferację komórek czerniaka oraz ich inwazyjny charakter. Promują również proces angiogenezy oraz zjawisko immunosupresji. Podobną rolę odgrywają makrofagi, które dodatkowo tworząc z komórkami czerniaka komórki hybrydowe nadają im bardziej agresywny fenotyp. Niezbędne w procesie rozsiewu są również komórki śródbłonkowe współtworzące nowe naczynia krwionośne. Powyższym procesom sprzyja niskie ciśnienie parcjalne tlenu w skórze, pogłębiające się w miarę rozwoju nowotworu. Niewykluczone, że sprzyjające rozwojowi czerniaka mikrośrodowisko jest jednym z czynników warunkujących jego niezwykle agresywny wzrost.
Słowa kluczowe:czerniak skóry • mikrośrodowisko nowotworowe • hipoksja
Summary
Melanoma develops from melanocytes localized mainly in skin, to a lesser extent in uvea and mucosal tissue. It is one of the most aggressive tumors, resistant to standard therapies. It is caused mainly by UV radiation. Approximately 10% of melanomas have a familial background. Transformation of melanocytes is driven mainly by mutations in genes coding for proteins regulating proliferation, cell growth, cell cycle, and death. Although genetic changes are indisputable causes of melanoma formation, the key role of the skin microenvironment is becoming more and more acknowledged.
Normal cells of the skin, such as keratinocytes, fibroblasts, endothelial, and immune cells, alongside melanocytes constitute a special microenvironment in which disturbed homeostasis may facilitate melanoma development. The process of transformation is initiated by aberrant interaction of keratinocytes and melanocytes. Proliferation and invasive growth of the developing neoplasm may be further facilitated by myofibroblasts as well as macrophages residing within the tumor. They are able to stimulate angiogenesis and suppress the immune response. Macrophages may, additionally, create aggressive hybrid cells through fusion with melanoma cells. Indispensable in the process of metastasis are endothelial cells which co-form blood vessels, the main routes of tumor cells’ seeding. All of the aforementioned processes take place in a tumor growth-promoting atmosphere of low oxygen tension in the skin, further decreasing during tumor growth. Probably the tumor growth-promoting microenvironment is one of the main factors responsible for melanoma aggressiveness.
Key words:skin melanoma • tumor microenvironment • hypoxia
Wykaz skrótów:
ADAM-9 – domena 9 metalopeptydazy ADAM (ADAM metallopeptidase domain 9), PAR-1 – receptor aktywowany proteinazą 1 (protease-activated receptor-1), AKT/PKB – kinaza białkowa B (protein kinase B), Alpha-MSH – hormon stymulujący melanocyty (melanocyte stimulating hormone), ANG-2 – angiogenina 2 (angiogenin 2), ANXA2 – aneksyna A2 (annexin A2), BCL-2 – regulator apoptozy należący do rodziny białek BCL (B-cell CLL/lymphoma 2), bFgf2 – (zasadowy) czynnik wzrostu fibroblastów ((basic) fibroblast growth factor), Hgf – czynnik wzrostu hepatocytów (hepatocyte growth factor), BNIP3L – białko wiążące Bcl-2 i adenowirusowe białko E1B-19 Da 3 (BCL2/adenovirus E1B 19 kDa interacting protein 3-like), BRAF – protoonkogenna kinaza serynowo-treoninowa, homolog wirusowego onkogenu v-raf mięsaka mysiego (v-raf murine sarcoma viral oncogene homolog B1), CAF – fibroblasty zasiedlające nowotwory (cancer-associated fibroblasts), CAIX – anhydraza węglanowa IX (carbonic anhydrase IX), CCL – ligand dla chemokiny C-C (chemokine (C-C motif) ligand), CDC6 – białko 6 cyklu podziałowego komórki (cell division cycle 6), CXCL – ligand dla chemokiny z motywem C-X-C (chemokine (C-X-C motif) ligand), CDH5 – kadheryna 5, typ 2 (cadherin 5, type 2), COX-2 – cyklooksygenaza 2 (cyclooxygenase 2), CSF-1 – czynnik stymulujący tworzenie kolonii makrofagów (colony stimulating factor 1 (macrophage)), CXCR4/SDF – chemokina typu 4 / zrębowy czynnik wzrostu (chemokine (C-X-C motif) receptor 4 / stromal derived factor), E-CAD – kadheryna E (cadherin E), EPC – komórki progenitorowe śródbłonka (endothelial progenitor cell), EPHA2 – 2 efrynowy receptor typu A (ephrin type-A receptor 2), ESAM – cząsteczka adhezyjna komórek śródbłonkowych (endothelial cell adhesion molecule), ET-1/ET(B) – endotelina 1 i jej receptor typu B (Endothelin-1/ETB Receptor), FN – fibronektyna (fibronectin), GLUT1 – transporter glukozy 1 (Glucose transporter 1), GMC-SF – czynnik stymulujący tworzenie kolonii granulocytów i makrofagów (granulocyte macrophage colony-stimulating factor), HIF-1-2 – czynnik indukowany hipoksją 1, – 2 (hypoxia-inducible factor-1, – 2), HPV8 – wirus brodawczaka ludzkiego typu 8 (human papillomavirus type 8), ICAM-1 – międzykomórkowa cząsteczka adhezyjna 1 (intercellular adhesion molecule 1), IL-6 – interleukina 6 (interleukin 6), Il-8 – interleukina 8 (interleukin 8), IL-beta – interleukina beta (interleukin beta), JAMs – cząsteczki adhezyjne typu „przylegania”(junctional adhesion molecules), KGF – czynnik wzrostu keratynocytów (keratinocyte growth factor), LAMP-1 – lizosomalne białko błonowe 1 (lysosomal-associated membrane protein 1), LGALS3 – galektyna 3 (lectin, galactoside-binding, soluble, 3), LOX – oksydaza lizylowa (lysyl oxidase), LPPCN – „liniowa programowana śmierć nekrotyczna” (lineary patterned programmed cell necrosis), MAPK – kinazy aktywowane mitogenami (mitogen-activated protein kinases), MCAM – cząsteczka adhezyjna komórek czerniaka (melanoma cell adhesion molecule), MELAN-A/MTART1 – antygen czerniaka rozpoznawany przez limfocyty T (melanoma antigen recognized by T-cells 1), MITF – czynnik transkrypcyjny związany z (microphthalmia-associated transcription factor), MMPs – metaloproteinazy (metaloproteinases), MT1MMP – macierzowa metaloproteinaza 1, typ błonowy (membrane-type matrix metalloproteinase-1), MXI1 – białko wchodzące w interakcję z białkiem MAX (MAX interactor 1), N-CAD – kadheryna N (cadherin N), NK – komórki układu immunologicznego typu „naturalni zabójcy” (natural killer), NME1 – białko 1 ulegające ekspresji w komórkach nieprzerzutujących (non-metastatic cells 1, protein (NM23A) expressed in), NOTCH-1 – homolog 1 genu Notch (Notch gene homolog 1), NRAS – (neuroblastoma RAS viral (v-ras) oncogene homolog), PFKL – fosfofruktokinaza wątrobowa (phosphofructokinase, liver), PFKM – fosfofruktokinaza mięśniowa (phosphofructokinase, muscle), P21 – inhibitor 1 kinaz zależnych od cyklin (cyclin-dependent kinase inhibitor 1), PHD3 – hydroksylaza prolinowa (prolyl hydroxylase 3), PI3K – kinaza fosfatydyloinozytolu (Phosphoinositide 3-kinase), PLAU – urokinazowy aktywator plazminogenu (plasminogen activator, urokinase), RGP – faza wzrostu poziomego (radial growth phase), RXRalpha – receptor kwasu retinowego (retinoic acid receptor RXR-alpha), Scf – czynnik wzrostu komórek macierzystych (stem cell factor), SOCS-1 – supresor szlaku sygnalizacyjnego cytokiny 1 (suppressor of cytokine signaling 1), SPARC – osteonektyna (secreted protein, acidic, cysteine-rich (osteonectin)), STAT3 – transduktor sygnału i aktywator transkrypcji 3 (signal transducer and activator of transcription 3), TAMs – makrofagi zasiedlające nowotwory (tumour-associated macrophages), TEM – tranzycja (przejście) epitelialno-mezenchymalna, TGF-β – transformujący czynnik wzrostu, beta (transforming growth factor beta), PDGF-A – płytkopochodny czynnik wzrostu, alpha (platelet-derived growth factor alpha), TIL – limfocyty naciekające nowotwór (tumour infiltrating lymphocytes), TIM-3 – transbłonowa immunoglobulina i mucyna 3 (T cell immunoglobulin and mucin 3), TIMPs – tkankowe inhibitory metaloproteinaz (tissue inhibitors of metalloproteinases), TNF-alpha – czynnik martwicy nowotworów alpha (tumour necrosis factor alpha), TYR – tyrozynaza (tyrosinase), UV – promieniowanie ultrafioletowe (ultraviolet radiation), VCL – winkulina (vinculin), VEGF – czynnik wzrostu śródbłonka naczyniowego (vascular endothelial growth factor), VGP – faza wzrostu pionowego (vertical growth phase), VIM – wimentyna (vimentin), WISP-1 – białko indukowane przez WNT1 (WNT1-inducible-signaling pathway protein 1).
Wstęp
Zmiany genetyczne, epigenetyczne i funkcjonalne w komórkach nowotworowych nie są jedynymi czynnikami procesu kancerogenezy. Istotna, coraz lepiej poznawana, jest rola mikrośrodowiska nowotworowego. Mikrośrodowisko nowotworowe to strukturalna i funkcjonalna konstelacja komórek nowotworowych i nienowotworowych, wzajemnie na siebie oddziałujących przez kontakt oraz zewnątrzkomórkowe substancje aktywne, takie jak czynniki wzrostu, cytokiny, metaloproteazy i inne białka [72,116]. Definicję tę można uzupełnić o jeszcze jeden, nieodłączny element mikrośrodowiska nowotworowego, jakim jest stan niedotlenowania, czyli hipoksja.
Czerniak jest jednym z nowotworów, którego powstanie i rozwój jest ściśle powiązany z mikrośrodowiskiem skóry, w którym się znajduje. O tym, czy prawidłowy melanocyt przekształci się w komórkę nowotworową, która da początek rozwijającemu się czerniakowi, nie decydują tylko mutacje powstałe w jego DNA, ale również wpływ otaczających go komórek. Jednocześnie następstwem nieprawidłowości w materiale genetycznym melanocytów jest zaburzona relacja z tymi komponentami skóry, czyli keratynocytami, fibroblastami, komórkami śródbłonka oraz komórkami układu immunologicznego. W ten sposób zmiany molekularne zachodzące w komórkach barwnikowych oraz zależne lub niezależne od nich funkcjonowanie komórek prawidłowych sprzyjające procesom nowotworowym razem promują proces rozwoju czerniaka. Dogodne warunki tym patologicznym zmianom tworzy stan niedotlenowania występujący w skórze pogłębiający się w miarę wzrostu nowotworu.
Rola komórek prawidłowych wpowstaniu i progresji czerniaka
Keratynocyty
Czerniak wywodzi się z komórek barwnikowych, melanocytów, umiejscowionych w warstwie podstawnej naskórka oraz błonach śluzowych. Melanocyty wytwarzają barwnik, melaninę, który następnie za pomocą wypustek dendrytycznych transportują do keratynocytów, gdzie chroni on DNA komórek skóry przed szkodliwym działaniem promieniowania UV. Jeden melanocyt „obsługuje” około 30-35 keratynocytów, z którymi tworzy tzw. naskórkową jednostkę melaninową (epidermal melanin unit) cechująca się liczebną równowagą tych dwóch typów komórek występujących w stosunku 1 melanocyt na 35 keratynocytów [78]. Proliferacja melanocytów zachodzi podczas wzrostu skóry w okresie dzieciństwa oraz w organizmie dorosłym pod wpływem bodźców, takich jak promieniowanie słoneczne lub zranienia. Proces ten przebiega kilkuetapowo. Pierwszy etap to zerwanie fizycznego kontaktu (decoupling) melanocytów z keratynocytami. W następnym etapie komórki dzielą się i przemieszczają wzdłuż warstwy podstawnej naskórka. W końcowym etapie melanocyt nawiązuje kontakt z otaczającymi go keratynocytami. Cały proces jest pod ścisłą kontrolą keratynocytów. Zaburzenie homeostazy jednostki melaninowej w wyniku zerwania kontaktu między tymi dwoma typami komórek może spowodować niekontrolowane podziały melanocytów niezależnie od molekularnych sygnałów płynących z keratynocytów. Wykazano, że melanocyty hodowane bez obecności keratynocytów zawierają na powierzchni komórki antygeny związane z czerniakiem, takie jak białko adhezyjne, MCAM [147] czy integryna αvβ3, będące głównymi regulatorami inwazyjnego wzrostu czerniaka [53]. Konsekwencją uniezależnienia się melanocytów od keratynocytów jest zatem ich niekontrolowany podział, który stanowi pierwszy etap transformacji nowotworowej [39,80] i rozwoju czerniaka [25].
Uniezależnienie się melanocytów od keratynocytów jest spowodowane zaburzeniem kontaktu między tymi dwoma typami komórek. Główną rolę w tej komunikacji odgrywają białka adhezji komórkowej, spośród których najistotniejszą grupę stanowią kadheryny, zwłaszcza kadheryna E [133]. Zmniejszenie, brak ekspresji lub obecność niefunkcjonalnej E-kadheryny obserwuje się w większości czerniaków i jest ono związane z jego inwazyjnym wzrostem [39]. Przywrócenie funkcji tej kadheryny w komórkach czerniaka zwiększa ich wrażliwość na apoptozę [63], zmniejsza ilość białek inwazyjnych na powierzchni komórek, przywraca kontrolę wzrostu przez keratynocyty [52] oraz hamuje ich proliferację [79]. Komórka melanocytu może utracić E-kadherynę w wyniku wielu mechanizmów molekularnych. Należą do nich mutacja w genie kodującym E-kadherynę (CDH1) lub beta-kateninę, nadekspresja supresorów tego białka, np. czynników transkrypcji Snail, Slug, [78], aktywacja białka NOTCH-1 [101], a także aktywacja ścieżek sygnałowych MAPK, PI3K lub Rho [23,39]. Powyższe zmiany mogą być indukowane również przez mikrośrodowisko melanocytów, w tym przez same keratynocyty.
Jaki jest mechanizm tego procesu? Keratynocyty wydzielają czynniki, które regulują ekspresję E-kadheryny na powierzchni melanocytów. Obserwowano np. obniżenie ekspresji CDH1 pod wpływem czynnika wzrostu, endoteliny 1 (ET-1). Aktywacja ścieżki ET-1/ET(B) w keratynocytach następuje pod wpływem działania promieniowania UVB i prowadzi do obniżenia ekspresji E-kadheryny oraz związanych z nią katenin w komórkach melanocytów i czerniaka [59]. Niewykluczona jest również rola jądrowego receptora RXRalpha. Utrata ekspresji tego białka w keratynocytach koreluje z progresją czerniaka. Ponadto, autorzy pracy zaobserwowali, że komórki czerniaka z mutacją Cdk4 hodowane razem z keratynocytami pozbawionymi tego białka, łatwiej przerzutowały do węzłów chłonnych. Powstałe guzy wtórne cechowała obniżona ekspresja supresorów nowotworowych Pten, Fas i właśnie E-kadheryny, natomiast zasiedlające guzy keratynocyty wytwarzały czynniki wzrostu: Fgf2, Hgf, Scf, α-MSH i Et-1, [56]. Niewykluczone, że keratynocyty wpływają na proliferację melanocytów również przez regulację ekspresji E-kadheryny na powierzchni swoich własnych komórek. Turner i wsp. [136] wykazali, że pod wpływem czynnika transkrypcyjnego Slug, wytwarzanego w keratynocytach, zmniejsza się ekspresja CDH1 na ich powierzchni, co prowadzi do obniżenia proliferacji keratynocytów. Wykazano również, że promieniowanie UVB indukuje śmierć apoptotyczną keratynocytów [95]. Wszystkie opisane procesy mogą się przyczynić do zachwiania homeostazy jednostki melaninowej, a w konsekwencji do ograniczenia kontroli podziałów melanocytów przez keratynocyty.
Innym sposobem, w jaki keratynocyty mogą wpływać na powstanie i rozwój czerniaka jest wydzielanie czynników wzrostu. Czynnik SCF wytwarzany przez keratynocyty zwiększa potencjał proliferacyjny komórek czerniaka w wyniku aktywacji ścieżki SCF/C-KIT. Co ciekawe, wydzielanie SCF przez keratynocyty może być następstwem działania innego czynnika wzrostu, KGF, wydzielanego przez fibroblasty [9]. W ten sposób tworzy się sieć parakrynnych zależności między poszczególnymi elementami komórkowymi mikrośrodowiska nowotworowego. Inne czynniki wydzielane prze keratynocyty to wspomniana wyżej endotelina 1, a także Hgf i α-MSH [56].
Oprócz nadzorowania proliferacji melanocytów, keratynocyty uczestniczą również w promowaniu ich inwazyjnego fenotypu. Badania na modelu „zrekonstruowanej skóry” wykazały, że obecność keratynocytów w skórze jest konieczna do inwazyjnego wzrostu komórek czerniaka. Komórki czerniaka o inwazyjnym fenotypie miały zdolność przechodzenia przez błonę podstawną tylko wtedy, gdy były hodowane w obecności keratynocytów. Mediatorem procesu była metaloproteinaza MMP9 aktywowana przez komórki nowotworowe w obecności keratynocytów [137]. Mechanizm tego zjawiska nie jest znany. Być może w procesie indukcji ekspresji metaloproteinaz uczestniczą cytokiny prozapalne wydzielane przez keratynocyty pod wpływem UV. Wykazano np., że TNF-alfa i IL-beta aktywują MMP9 w komórkach czerniaka [125]. Niewykluczona jest również rola białek wirusowych. Obserwowano aktywację promotora metaloproteinazy MMP9 w keratynocytach pod wpływem białka E2 wirusa HPV8 [1]. Rolę metaloproteinaz wydzielanych przez komórki nienowotworowe w rozwoju czerniaka potwierdzają również wyniki badań Müllera i wsp. [90]. Autorzy zaobserwowali, że wraz z progresją czerniaka rośnie ekspresja MMP19 w keratynocytach i fibroblastach sąsiadujących z komórkami nowotworowymi. Zainteresowanych rolą tych białek w rozwoju czerniaka odsyłam do pracy Fröhlicha [34]. Schemat roli keratynocytów w rozwoju czerniaka przedstawia ryc. 1.

Ryc. 1. Rola keratynocytów w powstaniu i progresji czerniaka. Proliferacja melanocytów znajduje się pod kontrolą keratynocytów, z którymi są w ścisłym kontakcie. Mediatorem tego kontaktu są białka adhezyjne, głównie kadheryna E. Czynniki wzrostu wydzielane przez keratynocyty np. pod wpływem promieniowania UV lub stymulowane białkami wydzielanymi przez miofibroblasty (KGF) prowadzą do obniżenia ekspresji E kadheryny na powierzchni melanocytów oraz stymulacji ich proliferacji. Podobny efekt daje zmniejszenie ekspresji E-kadheryny na powierzchni keratynocytów pod wpływem działania czynnika transkrypcyjnego SLUG oraz zmniejszenie ekspresji czynnika RXR?. Poprzez wydzielanie lub stymulowanie wydzielania metaloproteinaz keratynocyty uczestniczą również w procesie inwazji czerniaka. Niewykluczona jest tutaj rola wirusa HPV8
Zmniejszeniu ekspresji E-kadheryny na komórkach czerniaka zazwyczaj towarzyszy wzrost ekspresji N-kadheryny. Zmiana profilu ekspresji tych dwóch typów kadheryn (cadherin switching) powoduje zaburzenia interakcji melanocytów/komórek czerniaka z sąsiadującymi komórkami prawidłowymi. Przerwany zostaje kontakt z keratynocytami, natomiast komórka nabywa zdolność interakcji z fibroblastami i komórkami endotelialnymi, co jest istotnym zdarzeniem w nabyciu inwazyjnego fenotypu przez te komórki. Proces ten jest regulowany również przez same fibroblasty, o których mowa poniżej.
Fibroblasty
Fibroblasty są kolejnym istotnym komponentem komórkowym mikrośrodowiska nowotworowego (cancer associated fibroblasts, CAFs). Pełnią kilka zasadniczych funkcji: promują wzrost nowotworu przez wydzielanie czynników wzrostu, regulują proces angiogenezy, organizują podścielisko wytwarzając komponenty macierzy zewnątrzkomórkowej (kolagen, fibronektyna, laminina); jednocześnie wydzielając kolagenazę uczestniczą w rozkładzie macierzy pozakomórkowej, co jest niezbędne w procesie przemieszczania się komórek i tworzenia przerzutów [96,116,139].
Na początkowym etapie rozwoju czerniaka, w fazie RGP (radial growth phase), gdy zmiana nie ma cech inwazyjnych, fibroblasty mogą hamować dalszy rozwój tego nowotworu [26]. Wykazano również, że fibroblasty z aktywną ścieżką NOTCH1 hamują wzrost czerniaka, a pośrednikiem tego procesu jest białko WISP-1 [124]. Podczas dalszego rozwoju nowotwór w kilkuetapowym procesie, opisanym dokładnie przez Ruitera i wsp. [116] „przeciąga na swoją stronę” fibroblasty. W pierwszym etapie, pod wpływem czynników wzrostu, takich jak np. bFGF, TGF-β czy PDGF-A, fibroblasty rezydujące, ich prekursory lub komórki macierzyste przemieszczają się w kierunku komórek nowotworowych. Następnie odbywa się aktywacja i proliferacja fibroblastów. W ostatnim etapie ulegają przekształceniu do miofibroblastów lub różnicowaniu do fibrocytów [116]. W zależności od pełnionych funkcji i umiejscowienia w guzie autorzy wyróżniają cztery typy fibroblastów: rezydujące, aktywowane, miofibroblasty i zróżnicowane fibroblasty. Fibroblasty rezydujące są umiejscowione w sąsiadującej tkance zdrowej i mogą być w każdej chwili „rekrutowane” przez nowotwór. Fibroblasty aktywowane rezydują w bezpośrednim sąsiedztwie nowotworu, gdzie są gotowe do podziałów i różnicowania w inne komponenty komórkowe (miofibroblasty, perycyty, fibrocyty), jednak nie są całkowicie zaprzęgnięte na użytek nowotworu. Kluczowe znaczenie dla funkcjonowania guza mają miofibroblasty oraz zróżnicowane fibroblasty. Te pierwsze znajdują się w rejonie czoła inwazyjnego (leading edge), gdzie przez wydzielanie czynników wzrostu oraz rozkład macierzy zewnątrzkomórkowej pod wpływem proteaz, promują wzrost i inwazję nowotworu. Zróżnicowane fibroblasty, rezydujące wewnątrz guza pełnią funkcję swoistego rusztowania dla masy nowotworowej [80,116]. Główne czynniki wzrostu wydzielane przez miofibroblasty to IGF-1, SC (scatter factor), bFGF, TGF-β i endoteliny. Białka te parakrynnie pobudzają komórki nowotworowe do wzrostu i proliferacji. Jednocześnie czynniki o podobnych funkcjach wydzielane przez komórki nowotworowe (np. wspomniany wyżej PDGF) stymulują podziały fibroblastów. Być może oba procesy napędzają się w wyniku dodatniego sprzężenia zwrotnego prowadząc do szybkiego wzrostu czerniaka [116,139].
Oprócz napędzania proliferacji komórek czerniaka, niektóre czynniki wzrostu wydzielane przez fibroblasty, np. TGF-β, stymulują komórki nowotworowe do wydzielania białek niezbędnych w procesie inwazji i przerzutowania, np. kolagenu czy tenascyny [78]. Dodatkowo HGF, powszechnie znany jako czynnik wydzielany przez fibroblasty, reguluje ekspresję dwóch podstawowych w transformacji i nabywaniu inwazyjnego fenotypu białek adhezyjnych, E- i N-kadheryny, powodując zmniejszenie ekspresji tej pierwszej i podwyższenie ekspresji drugiej (cadherin shift) [66]. Fibroblasty wydzielają również proteazy rozkładające komponenty macierzy zewnątrzkomórkowej, głównie kolagen I. Najbardziej znane to białka z grupy metaloproteinaz (MMP1) [140] oraz proteaza serynowa FAP (fibroblast activation protein). FAP jest głównym białkiem sprzyjającym progresji nowotworów wydzielanym przez fibroblasty. Korelację nadekspresji FAP z gorszą prognozą dla pacjentów obserwuje się w wielu typach nowotworów [106]. W czerniaku dane dotyczące tego białka są niejednoznaczne. Z jednej strony FAP wydzielany przez fibroblasty pod wpływem promieniowania UV zwiększa inwazyjność komórek czerniaka oraz reguluje ekspresję białka Fas [142]. Z drugiej strony, Ramirez-Montagut i wsp. [109] obserwowali obniżenie tumorogenności komórek czerniaka, w których eksperymentalnie uzyskano nadekspresję białka FAP. Niewykluczone, że protumorogenne właściwości FAP są związane z ekspresją tego białka przez fibroblasty, natomiast jego ekspresja w komórkach czerniaka skutkuje przeciwnym efektem. Spekulacje te są zgodne z obserwacjami, że FAP ulega ekspresji głównie w fibroblastach oraz w melanocytach niektórych znamion, natomiast nie występuje w komórkach czerniaka pierwotnego i przerzutach [55] lub występuje tylko jedna jego podjednostka [110].
Ważnym mediatorem wzrostu czerniaka wytwarzanym przez fibroblasty jest osteopontyna, OPN [17]. Pod wpływem czynnika wzrostu PDGF-CC wydzielanego przez komórki czerniaka fibroblasty nie tylko migrują w rejon nowotworu, ale również wydzielają osteopontynę – glikoproteinę odpowiedzialną za indukcję angiogenezy i proces przerzutowania w wielu typach nowotworów [89]. Ze względu na udokumentowany wpływ na inwazję i przerzutowanie czerniaka, OPN jest potencjalnym markerem progresji tego nowotworu (więcej w [17]). Inne białko promujące za pośrednictwem fibroblastów inwazyjny fenotyp czerniaka to kaweolina 1 (Cav-1) [37]. Podobnie jak OPN i FAP, białko to reguluje organizację macierzy zewnątrzkomórkowej. Na istotną rolę kaweoliny w rozwoju czerniaka wskazuje fakt, że przerzuty czerniaka są wzbogacone w fibroblasty wytwarzające to białko.
Oprócz wydzielania czynników wzrostu i proteaz rozkładających macierz zewnątrzkomórkową, fibroblasty regulują fenotyp komórek czerniaka wchodząc z nimi w bezpośredni kontakt. Jednym z mediatorów tych interakcji jest białko ADAM-9 [148]. To adhezyjne białko o właściwościach proteolitycznych umożliwia bezpośredni kontakt tych dwóch typów komórek sprzyjając w ten sposób inwazyjności komórek czerniaka. Ponadto, fibroblasty wraz z innymi komórkami (śródbłonek, makrofagi, płytki krwi) wydzielają również białko PAR-1, które jest zaangażowane w proces stanu zapalnego, koagulacji i pełni dobrze udokumentowaną rolę w procesie przerzutowania [146].
W dalszym wzroście guza fibroblasty uczestniczą również poprzez indukcję procesu angiogenezy, czyli tworzenia nowych naczyń krwionośnych. Wydzielany przez nie IGF-1 stymuluje wytwarzanie czynników proangiogennych (IL-8, VEGF) przez komórki nowotworowe. Fibroblasty mają również zdolność różnicowania się do perycytów i strukturalnie mogą brać udział w budowie nowych naczyń [80]. Uczestniczą również w zjawisku immunosupresji, czyli ucieczki komórek nowotworowych spod nadzoru układu immunologicznego. Wykazano, że fibroblasty mogą hamować przeciwnowotworową aktywność komórek NK przez zmniejszenie na powierzchni tych komórek receptorów niezbędnych do rozpoznania i eliminacji komórek czerniaka. Mediatorem tego procesu jest prostaglandyna wydzielana przez aktywowane fibroblasty [4].
Dotychczasową wiedzę o roli fibroblastów w progresji czerniaka próbuje się wykorzystać opracowując nowe strategie terapeutyczne nacelowane na ten komponent nowotworu, np. wykorzystując białka swoiste dla fibroblastów. Lee i wsp. [77] obserwowali przeciwnowotworowy efekt immunoterapii z wykorzystaniem przeciwciał swoiście rozpoznających białko FAP; szczepionka anty-FAP skutecznie hamowała wzrost zarówno guzów czerniaka (B16/F10.9), raka sutka (4T1) oraz grasiczaka (EL4). Schemat roli fibroblastów w rozwoju czerniaka przedstawia rycina 2.

Ryc. 2. Rola fibroblastów w powstaniu i rozwoju czerniaka. Fibroblasty stymulują wzrost komórek czerniaka poprzez wydzielanie czynników wzrostu (HGF, IGF, bFGF, SC, TGF-β). Ponadto ułatwiają inwazję poprzez obniżenie ekspresji E-kadheryny i podwyższenie ekspresji N-kadheryny na powierzchni melanocytów/komórek czerniaka, wydzielanie białek rozkładających macierz zewnątrzkomórkową (MMP-1, FAP, OPN, CAV-1) oraz indukowanie wydzielania czynników proangiogennych (IL-8, VEGF). W promocji czerniaka uczestniczą również inne białka wytwarzane przez fibroblasty, takie jak ADAM9, czy PAR-1. Fibroblasty upośledzają również działanie komórek NK przez wydzielanie prostaglandyny
Komórki śródbłonkowe i perycyty
Istotnym z punktu widzenia rozwoju nowotworu komponentem mikrośrodowiska są komórki śródbłonkowe (endotelialne) współtworzące naczynia krwionośne. Razem z naczyniami limfatycznymi, naczynia krwionośne stanowią główne drogi rozsiewu komórek nowotworowych do innych narządów. Stąd jednym z głównych procesów rozwoju guza angażujących mikrośrodowisko jest proces angiogenezy i limfogenezy. Tworzenie nowych naczyń krwionośnych i limfatycznych jest niezbędne do dalszego wzrostu i rozsiewu nowotworu, również czerniaka [5]. Angiogeneza w czerniaku jest autokrynnie i parakrynnie stymulowana przez czynniki wzrostu, proteazy oraz wiele czynników proangiogennych (dokładniej opisanych w [84]) wydzielanych przez komórki czerniaka (VEGF, bFGF, TGF-β, IL-8), a także prawidłowe komórki, np. fibroblasty (IGF-1) czy komórki endotelialne (IL-8). Źródłem czynników proangiogennych jest również macierz zewnątrzkomórkowa, z której czynniki te są uwalniane pod wpływem metaloproteinaz i ich inhibitorów wydzielanych zarówno przez komórki nowotworowe (MMP-1, – 2, – 9, – 13, – 14), jak i fibroblasty (MMP2, MT1MMP, TIMP1 i TIMP3 [116] oraz endotelium [139].
Warunkiem rozsiewu komórek nowotworowych jest ich przedostanie się do krwiobiegu. Na tym etapie komórki muszą mieć zdolność „przeciskania się” pomiędzy komórkami endotelialnymi. Proces ten jest uwarunkowany obecnością swoistych białek adhezyjnych na powierzchni komórek nowotworowych i endotelium. Jedną z grup białek pełniących tę funkcję są białka JAMs (junctional adhesion molecules [35,74]). Obecność białka JAM-C na powierzchni komórek czerniaka ułatwia ich migrację poprzez warstwę komórek endotelialnych, zaś po wędrówce komórek czerniaka z krwią do dalszych organów umożliwia im wyjście z naczyń, tzw. ekstrawazację, co w efekcie powoduje rozsiew czerniaka. [74]. Inną grupą nowo odkrytych białek zaangażowanych w nasilanie rozsiewu czerniaka są efryny. Wykazano, że ekspresja białka EphB4 na powierzchni komórek czerniaka sprzyja adhezji tych komórek do komórek endotelialnych wytwarzających ligand efrynę B2. Interakcja ta ułatwia rozsiew komórek czerniaka do specyficznych narządów (site-specific metastatic dissemination), np. płuc, nerek, wątroby [48]. Obecność efryny EPHA2 (jak również lamininy 5γ2 i VE-kadheryny) jest natomiast związana ze zdolnością komórek czerniaka do mimikry naczyniowej. Efryna EPHA2 prawdopodobnie ułatwia interakcję komórek czerniaka z komórkami endotelialnymi. Więcej o plastyczności komórek czerniaka pisze Hendrix i wsp. [47]. Innymi potencjalnie ważnymi białkami modulującymi interakcje komórek nowotworowych i endotelialnych są TIM-3 [144] oraz ESAM [20]. TIM-3 to białko pierwotnie rozpoznane jako receptor komórek Th1, który ulega ekspresji w komórkach endotelialnych i odpowiada za ich interakcje z komórkami czerniaka. Jego obecność ułatwia intrawazację, zwiększa przeżycie komórek czerniaka w świetle naczynia oraz moduluje ich ekstrawazację w miejscu przerzutu. Białko ESAM, należy do grupy immunoglobulin regulujących interakcje między komórkami endotelialnymi. Stymuluje migrację komórek endotelialnych i formowanie kapilar in vitro, natomiast in vivo reguluje proces przerzutowania [20]. Jeśli dalsze badania potwierdzą rolę tych białek w przerzutowaniu czerniaka mogą się one stać potencjalnym celem terapeutycznym w terapii uzupełniającej u pacjentów z wysokim ryzykiem nawrotu choroby. Schemat interakcji komórek endotelialnych i perycytów z komórkami czerniaka przedstawia rycina 3.

Ryc. 3. Rola komórek śródbłonkowych i perycytów w rozwoju czerniaka. Komórki endotelialne współtworzą naczynia krwionośne będące, obok naczyń limfatycznych, głównymi drogami rozsiewu czerniaka. Komórki czerniaka wydzielają czynniki proangiogenne regulujące proces angiogenezy oraz metaloproteinazy, które rozkładając macierz zewnątrzkomórkową ułatwiają migrację komórek oraz uwalniają zasoby czynników stymulujących proces neowaskularyzacji. Na komórkach zarówno czerniaka jak i śródbłonku znajdują się białka ułatwiające interakcje tych dwóch typów komórek w procesie rozsiewu (EPHB4, TIM-3, ESAM, JAM-C), a także regulujące proces mimikry naczyniowej (EPHA2). W stabilizacji unaczynienia guza biorą udział również perycyty
Interesujące są wyniki, które sugerują, że komórki endotelialne mogą również przeciwdziałać procesowi rozsiewu. Na modelu zwierzęcym zaobserwowano pochłanianie komórek czerniaka B16 przez prekursorowe komórki endotelialne. Proces ten był zależny od interakcji tych dwóch typów komórek regulowanej przez białko SPARC. Co ciekawe, tylko komórki EPC stymulowane przez komórki nowotworowe miały zdolność pochłaniania komórek nowotworowych [30].
Ważnym elementem mikrośrodowiska okołonaczyniowego i potencjalnym celem terapeutycznym są również perycyty – komórki otaczające naczynia włosowate. Aktywowane perycyty nowotworów częściej się dzielą i są zdolne przekształcić się nie tylko w perycyty, ale również adipocyty, komórki mięśni gładkich naczyń czy komórki tworzące macierz zewnątrzkomórkową [46]. Wykazano, że interakcje komórek endotelialnych i perycytów angażujące ścieżkę sygnałową PDGF są kluczowe w utrzymywaniu stabilizacji nowotworowych naczyń krwionośnych, a tym samym wzrostu guza [46]. Rolę perycytów w rozwoju czerniaka wykazano na modelu myszy pozbawionych białka NG2 (nerve/glial antigen 2), będącego istotnym komponentem perycytów. U myszy tych obserwowano zmniejszoną waskularyzację i ograniczony wzrost czerniaka oka [98]. Terapia nacelowana na tę grupę komórek mikrośrodowiska nowotworowego powodowała zmniejszenie guza w wyniku jednoczesnego działania czynników hamujących receptor PDGF na perycytach oraz VEGFR na komórkach endotelialnych [43]. Natomiast Nisancioglu i wsp. [92] nie wykazali zwiększonej skuteczności terapii antyangiogennej różnych typów nowotworów, w tym czerniaka mysiego B16, pozbawionych perycytów.
Odkąd odkryto rolę naczyń krwionośnych i procesu angiogenezy w rozwoju nowotworów, w tym czerniaka, zarówno naczynia krwionośne, jak i sam proces neowaskularyzacji stały się obiecującym celem terapeutycznym. Podstawowym celem tych terapii jest zahamowanie ścieżki głównego regulatora angiogenezy, VEGF. Podczas gdy w pewnych typach nowotworów, np. raku nerki, monoterapia z wykorzystaniem przeciwciała rozpoznającego VEGF (bewacyzumab) okazała się wystarczająco skuteczna, a w innych (rak piersi, płuca, jelita grubego, rak nerki z przerzutami, glejak) zwiększała skuteczność chemioterapii, to w czerniaku większość przeprowadzonych prób klinicznych nie wykazała większej skuteczności terapeutycznej, zarówno w monoterapii, jak i terapii kombinowanej [27]. Mała skuteczność terapii antyangiogennych w czerniaku może wynikać z tego, że angiogeneza nie jest jedynym procesem tworzenia naczyń krwionośnych w tym nowotworze. W tzw. procesie kooptowania, czerniak może wykorzystywać do własnych celów istniejące okoliczne unaczynienie, a w procesie mimikry naczyniowej jest zdolny tworzyć kanały rozsiewu komórek nowotworowych bez udziału komórek endotelialnych [117]. Więcej na temat angiogenezy, unaczynienia, ich roli w progresji czerniaka oraz możliwości wykorzystania w strategiach terapeutycznych piszą Helfrich i Schadendorf [46].
Na proces angiogenezy i limfogenezy wpływają nie tylko czynniki związane z mikrośrodowiskiem nowotworowym, ale również procesy, które dzieją się w innych częściach organizmu, jak np. rozwijająca się ciąża. Analiza czynników spoza mikrośrodowiska nowotworowego wykracza poza ramy tej pracy, dlatego przytoczę tylko podstawowe fakty dotyczące tej kwestii. Większość dotychczasowych analiz epidemiologicznych nie wykazała wpływu ciąży i statusu hormonalnego kobiety na rozwój czerniaka [38], jednak nie brakuje opisów przypadków, które sugerują związek między ciążą [118,28,100] lub stosowaną terapią hormonalną [19] a rozwojem czerniaka. Również najnowsze badania na modelu mysim wskazują, że ciąża stymuluje wzrost guzów, tworzenie przerzutów oraz zwiększa śmiertelność myszy obarczonych czerniakiem B16. Prawdopodobną przyczyną jest intensywny rozwój naczyń krwionośnych i limfatycznych. Wykazano, że w porównaniu z grupą kontrolną guzy u myszy brzemiennych charakteryzowały się podwyższonym poziomem VEGF oraz intensywniejszą limfangiogenezą [61]. Niewykluczone, że czynniki hormonalne mogą indywidualnie wpływać na przebieg choroby, dlatego istotne wydaje się dalsze zgłębianie tego zagadnienia i poszukiwanie wyznaczników hormonowrażliwości tego nowotworu.
Czerniak rozsiewa się również drogą limfatyczną. Jednym z istotnych czynników prognostycznych jest obecność komórek nowotworowych w węźle wartowniczym, czyli pierwszym węźle, do którego spływa chłonka z okolic zmiany nowotworowej [85]. Naczynia limfatyczne nie tylko stanowią kanały rozsiewu komórek, ale również aktywnie przyciągają je do docelowych miejsc tworzenia przerzutów. Jednym z mechanizmów jest aktywacja ścieżki CXCR4/SDF. Komórki czerniaka z ekspresją białka CXCR4 umiejscawiają się w docelowych narządach, których naczynia limfatyczne wydzielają czynnik SDF-1 [62].
Komórki układu immunologicznego
Istotną rolę w procesie wzrostu nowotworu odgrywają również komórki układu odpornościowego. Jakkolwiek na początkowym etapie procesu nowotworzenia system immunologiczny może hamować progresję, to w dalszych etapach niektóre jego elementy sprzyjają rozwojowi nowotworu, uczestnicząc w kilku procesach: wzroście guza, angiogenezie oraz immunotolerancji. Spośród wszystkich komórek układu odpornościowego najlepiej poznana jest rola makrofagów. Naturalną funkcją makrofagów jest odpowiedź na infekcję oraz zranienia. Są pierwszymi komórkami, które pojawiają się w miejscach patologicznych zmian i, wydzielając cytokiny oraz chemokiny, przyciągają inne komórki immunologiczne. Wytwarzają również czynniki wzrostu i czynniki proangiogenne stymulujące proces gojenia się ran. W początkowym etapie rozwoju nowotworu makrofagi stanowią pierwszą linię obrony przed rozwijającym się nowotworem, jednakże z czasem zostają „przejęte” przez nowotwór i ich dotychczasowe funkcje sprzyjają jego progresji. W stanach patologicznych, np. w przewlekłym stanie zapalnym, mogą również inicjować proces nowotworzenia. Makrofagi, które niejako przeszły na stronę „wroga”, to tzw. makrofagi związane z nowotworem, TAMs (tumor-associated macrophages). Badania TAMs w różnych typach nowotworów wykazały, że ich rola w rozwoju nowotworu zależy od konstelacji cytokin wytwarzanych przez te komórki oraz czynnika CSF-1 (colony stimulating factor). Ekspresja cytokin, takich jak IL-4, – 12, – 14 czy GM-CSF, sprzyja przeciwnowotworowej aktywności makrofagów, natomiast obecność rozpuszczalnej postaci CSF-1 oraz IL-6 i TGF-β, powoduje, że komórki te są aktywnie przyciągane do guza lub w obszar stanu zapalnego. W miejscach przewlekłego stanu zapalnego nie tylko mogą inicjować proces nowotworzenia, ale również promować dalszy wzrost i progresję przez wydzielanie czynników wzrostu, stymulowanie angiogenezy, syntezę proteinaz i estrogenów, a także immunosupresję. W różnych typach nowotworów, między innymi raku piersi, szyjki macicy, prostaty czy glejakach, obecność makrofagów w guzie koreluje z rokowaniem pacjentów [103,108]. Jak makrofagi wpływają na rozwój czerniaka?
W czerniaku makrofagi zasiedlające nowotwór zwane są melanofagami ze względu na obecność melaniny pochodzącej od pochłoniętych komórek czerniaka. Makrofagi naciekające zmianę wykryto w różnych typach czerniaka: zmianach skórnych [11], w czerniaku oka [15,58] oraz błonach śluzowych nosa [126]. W powyższych badaniach obecność makrofagów korelowała z gorszą prognozą u pacjentów [15,58,126], aczkolwiek są również badania wykazujące dodatnią korelację obecności melanofagów w zmianie pierwotnej z prognozą dla pacjentów [40]. W zmianach skórnych makrofagi umiejscawiają się głównie w ogniskach pierwotnych, natomiast w mniejszym stopniu w przerzutach i znamionach. Charakteryzuje je ekspresja prozapalnego białka COX-2, które jest również wydzielane przez komórki nowotworowe [11]. Białko to jest dobrze udokumentowanym mediatorem stanu zapalnego promującego proces kancerogenezy w tkankach epitelialnych [122]. Makrofagi wydzielają również cytokiny regulujące podstawowe procesy w progresji nowotworu, takie jak rozsiew (TGF-β) [12], angiogeneza (IL-10) [21] czy immunotolerancja (IL-10) [29]. Jednocześnie brak lub zmniejszone wydzielanie niektórych cytokin może upośledzać procesy immunologiczne zachodzące w rozwijającym się czerniaku. Wykazano, że niska ekspresja CCL2, CCL3, CCL4, CCL5, CXCL9 oraz CXCL10 w zmianach nowotworowych czerniaka koreluje ze zmniejszonym naciekiem limfocytów [41]. Skądinąd wiadomo, że obecność limfocytów T w pierwotnej zmianie (TIL, tissue infiltrating lyphocytes) [24], a także komórek typu B [73] jest niezależnym, pozytywnym czynnikiem rokowniczym. Sprzymierzeńcem w walce z rozwijającym się nowotworem jest wspomniany wyżej GMC-SF. Najnowsze badania wskazują, że makrofagi stymulowane tą cytokiną wydzielają rozpuszczalną postać receptora VEGF (sVEGFR-1), która antagonizuje działanie VEGF. Proces jest mediowany przez czynnik HIF-2 i prowadzi u myszy do redukcji procesu angiogenezy [111]. Przeciwną rolę cytokin w progresji czerniaka sugerują badania roli białka SOCS-1 w rozwoju przerzutów czerniaka w mózgu [54]. SOCS-1 hamuje odpowiedź komórkową na różnego rodzaju cytokiny, takie jak np. IL-6, – 4. Wykazano, że zmniejszona ekspresja tego białka w przerzutach czerniaka do mózgu wiąże się z aktywacją ścieżki STAT3 oraz nadekspresją VEGF, bFGF oraz MMP-2, a w konsekwencji wzmożoną angiogenezą i inwazyjnością. Przywrócenie ekspresji tego białka, skutkuje natomiast znaczącym przyhamowaniem rozwoju przerzutów w mózgu.
Niewykluczone, że cytokiny prozapalne wydzielane przez komórki układu immunologicznego mogą się przyczyniać do powstania czerniaka. Między innymi, zaburzeniami w funkcjonowaniu układu immunologicznego, takimi jak podwyższony poziom cytokin prozapalnych i upośledzona aktywność limfocytów CD4, próbuje się tłumaczyć większą zachorowalność i gorsze rokowanie w przebiegu czerniaka u ludzi w starszym wieku [44]. Być może cytokiny prozapalne mogą się przyczyniać także do powstania czerniaka w wyniku mechanicznych urazów lub drażnienia znamion [135], aczkolwiek potwierdzenie tego przypuszczenia wymaga dalszych badań. Szerzej o roli cytokin w czerniaku pisze Navarini-Meury i wsp. [91].
Makrofagi mogą również bezpośrednio wpływać na powstanie agresywnego fenotypu czerniaka. Wykazano, że fuzja makrofagów z komórkami tego nowotworu prowadzi do powstania klonów komórkowych, które mają większą zdolność do chemotaksji, wyższy potencjał przerzutowania [76], większą ruchliwość oraz cechuje je aneuploidia i heterogenność [65]. Komórki hybrydowe charakteryzowała ekspresja kluczowych w progresji nowotworów białek regulujących takie procesy jak wiązanie komórek nowotworowych z endotelium (LAMP-1), czy przejście epitelialno-mezenchymalne (SPARC) [76]. Zdaniem autorów, zjawiska takie jak autofagia i efekt Wartburga, które są silnymi wyznacznikami nowotworowymi, mogą być skutkiem fuzji tych komórek z komórkami układu immunologicznego.
Układ immunologiczny ma również swój udział w procesie angiogenezy. Oprócz wspominanych proangiogennych cytokin wydzielanych przez komórki układu immunologicznego pewną rolę przypisuje się prekursorom komórek dendrytycznych. Haass i Herlyn [39] twierdzą, że mogą się one przekształcać w komórki endotelialne infiltrujące guz i uczestniczyć w powstawaniu nowych naczyń krwionośnych. Proces przerzutowania mogą promować również inne komórki układu immunologicznego, np. neutrofile polimorfojądrowe. Uczestniczą one w adhezji komórek czerniaka do komórek endotelialnych i ułatwiają w ten sposób ekstrawazację komórek nowotworowych w procesie rozsiewu [81]. Proces ten jest regulowany przez białka adhezyjne na komórkach czerniaka i neutrofilach, odpowiednio ICAM-1 i integrynę β. Co ciekawe, ekspresja integryny β jest stymulowana wydzielaną przez komórki nowotworowe cytokinę IL-8, o dobrze udokumentowanej roli w progresji czerniaka [86]. Schematyczne przedstawienie roli komórek układu immunologicznego w rozwoju czerniaka prezentuje rycina 4.
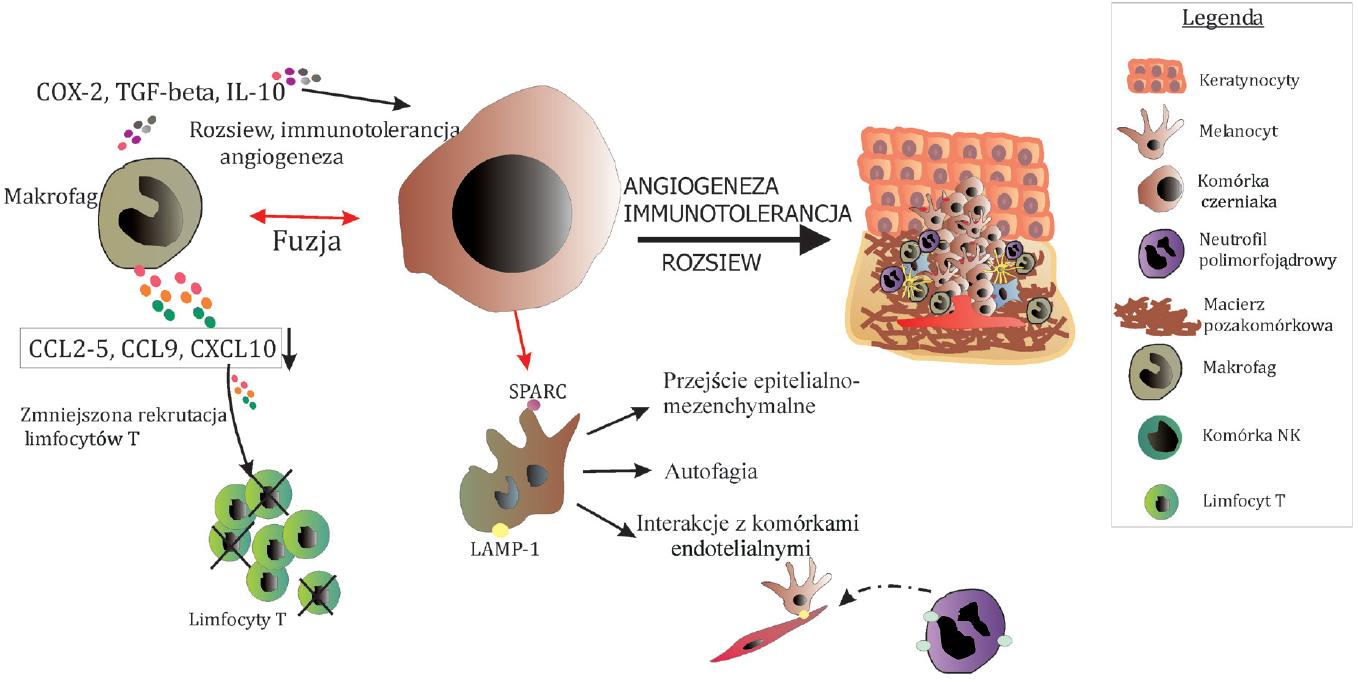
Ryc. 4. Rola komórek układu odpornościowego w rozwoju czerniaka. Makrofagi (melanofagi) wydzielają wiele czynników regulujących takie procesy jak angiogeneza (IL-10), przerzutowanie (TGF-beta) oraz immunosupresja (IL-10). Również zmniejszona ekspresja niektórych cytokin może prowadzić do upośledzenia rekrutacji limfocytów T w miejsce rozwijającego się nowotworu. Makrofagi mogą również ulegać fuzji z komórkami czerniaka tworząc komórki hybrydowe mające większą zdolność do autofagii, TEM i angiogenezy. Neutrofile natomiast regulują interakcje komórek czerniaka z endotelium podczas ekstrawazacji
Komórki czerniaka
W kontekście mikrośrodowiska nowotworowego nie można pominąć badań, które wskazują na istotny wpływ dojrzałych komórek nowotworowych na prawidłowe, niestransformowane melanocyty. Wykazano, że mikrośrodowisko złośliwych komórek czerniaka powoduje transróżnicowanie prawidłowych melanocytów do komórek złośliwych. Po czterodniowej inkubacji w trójwymiarowym mikrośrodowisku uprzednio prekondycjonowanym hodowlą komórek złośliwych, melanocyty nabywały bardziej inwazyjny fenotyp oraz wykazywały profil ekspresji zbliżony do profilu komórek złośliwych. Nadekspresji ulegały między innymi geny związane z procesem inwazyjności, takie jak np. EPHA2, VEGFC, PLAU, CDH5, VIM, natomiast bez zmian, na wysokim poziomie utrzymywała się również ekspresja genów związanych z różnicowaniem melanocytów (MITF, TYR, MELAN-A) [119]. Jeśli zjawisko to występuje również in vivo, takie epigenetyczne indukowanie inwazyjnego fenotypu w komórkach sąsiadujących z czerniakiem mogłoby częściowo tłumaczyć jego szybki wzrost i dużą agresywność.
Podsumowując, prawidłowe komórki mikrośrodowiska nowotworowego odgrywają istotną rolę na każdym etapie powstania i rozwoju czerniaka. Początkowo keratynocyty, makrofagi czy fibroblasty mogą działać przeciwnowotworowo, z czasem jednak rozwijający się nowotwór nabywa sprzymierzeńców w postaci miofibroblastów, melanofagów, perycytów oraz komórek układu immunologicznego. Wzajemna interakcja wszystkich komórkowych komponentów mikrośrodowiska czerniaka może sprzyjać jego szybkiemu i agresywnemu wzrostowi. Powyższe interakcje występują w warunkach niedotlenowania, które dodatkowo promuje rozwój czerniaka.
Rola hipoksji w powstaniu i progresji czerniaka
Rola hipoksji w procesie progresji różnych typów nowotworów jest bogato udokumentowana. Stan niedotlenowania wpływa na istotne dla rozwoju raka procesy, takie jak angiogeneza [31], immunosupresja [33], utrzymanie komórek macierzystych oraz wszystkie etapy procesu przerzutowania [83]. Coraz więcej danych wskazuje na ważną rolę hipoksyjnego mikrośrodowiska również w rozwoju czerniaka.
Podczas gdy w większości litych nowotworów stan niedotlenowania pojawia się dopiero na pewnym etapie wzrostu raka, gdy masa rosnącego guza jest niedostatecznie unaczyniona, w skórze łagodna hipoksja (3-5% O2) jest stanem naturalnym. Stąd od początku melanocyty i rozwijający się czerniak, niezależnie od wielkości, podlegają działaniu niskiego stężenia tlenu [32]. O stabilizacji czynnika HIF-1, głównego regulatora odpowiedzi komórek na stan niedotlenowania, w tych warunkach świadczy ekspresja w skórze podstawowych markerów hipoksji, takich jak CAIX i GLUT1 [6]. Stan hipoksji utrzymuje się na dalszych etapach rozwoju czerniaka zarówno w zmianach pierwotnych [42], jak i przerzutach [75].
Dopiero w ostatnich latach zaczęto poznawać i rozumieć znaczenie niedotlenowania w rozwoju czerniaka. Dotychczasowe badania wskazują, że obniżone stężenie tlenu może być jednym z istotnych czynników nie tylko promujących wzrost i progresję czerniaka, ale również inicjującym jego powstanie. Niewykluczone, że towarzyszący mu na każdym etapie rozwoju stan hipoksji lub fenotyp hipoksyjny komórek jest jednym z czynników odpowiedzialnych za dużą agresywność tego nowotworu.
Hipoksja a powstanie czerniaka
Badania ostatnich lat wskazują, że hipoksja może sprzyjać transformacji nowotworowej melanocytów. Jako pierwsi wyniki te zebrali i przedyskutowali pionierzy tych badań, Bedogni i Powell [6]. Ich zdaniem stan łagodnej hipoksji, który prowadzi do aktywacji ścieżki HIF-1, zapobiega starzeniu się melanocytów. W fizjologicznych (niepatologicznych) warunkach jest to korzystne zjawisko, ponieważ pozwala zachować pulę trudno odnawialnych komórek macierzystych melanocytów w skórze. Przeciwdziałanie starzeniu może jednak prowadzić jednocześnie do promowania transformacji nowotworowej melanocytów. Zdaniem autorów łagodna hipoksja (2%) indukując prożyciową ścieżkę HIF-1 sprzyja transformacji nowotworowej melanocytów przez blokowanie procesu starzenia melanocytów, które w wyniku aktywacji np. onkogenu BRAF mają przewagę proliferacyjną nad niezmutowanymi komórkami. W badaniach z 2005 roku, ci sami autorzy wykazali, że melanocyty z aktywną ścieżką AKT dużo łatwiej podlegają transformacji w warunkach hodowli przy obniżonym stężeniu tlenu niż w warunkach standardowych. Było to zależne od aktywności głównego regulatora odpowiedzi na hipoksję, czynnika HIF-1 [8]. Badania innych grup wykazały zależność kluczowych w rozwoju czerniaka białek, takich jak BRAF, NOTCH-1 czy MITF, z hipoksyjnym środowiskiem i aktywnością czynnika HIF-1. Zarówno onkogen BRAF, który jest najczęściej aktywowanym onkogenem w czerniaku [68], jak i MITF, często ulegający w czerniaku amplifikacji [18], stymulują ekspresję HIF-1 w melanocytach i w komórkach czerniaka. Aktywność HIF-1 w melanocytach ze zmutowanym BRAF zwiększa przeżycie tych komórek w warunkach hipoksji. Ponieważ mutacje BRAF wykrywa się również w znamionach, niewykluczone, że aktywacja ścieżki HIF-1 pod wpływem BRAF jest ważnym zdarzeniem w transformacji melanocytów, dającym tym komórkom przewagę nad niezmutowanymi w hipoksyjnym środowisku skóry.
Stan niedotlenowania stymuluje również ekspresję białka NOTCH-1 [7] pełniącego różnorakie funkcje w rozwoju nowotworów [115]. W czerniaku aktywacja NOTCH-1 występuje na wczesnym etapie transformacji melanocytów i warunkuje progresję tego nowotworu (więcej w [99]). Badania in vitro wykazały również, że obecność mutacji BRAF oraz aktywnej ścieżki Akt3 w melanocytach są wystarczające do transformacji tych komórek [22]. Czy czynnik HIF-1 jest mediatorem tego procesu wykażą dalsze badania. Niezależnie od udziału HIF-1 w powyższych regulacjach, dotychczasowe wyniki wskazują na ważną rolę ścieżki HIF-1 w promowaniu transformacji melanocytów, a najnowsze badania potwierdzają to przypuszczenie.
Wydaje się, że aktywność HIF-1 jest niezbędna również do transformacji melanocytów pod wpływem mutacji aktywującej białko c-Kit [88]. Melanocyty, w których ekspresji ulegało zarówno zmutowane białko c-Kit jak i czynnik HIF-1alfa miały zdolność tworzenia kolonii w agarze (wyróżnik transformacji nowotworowej). Komórki te cechowała konstytutywna aktywacja ścieżek PI3K/Akt oraz MAPK/ERK. Obecność w komórkach tylko zmutowanego białka c-Kit prowadząca do aktywacji ścieżki PI3K/Akt, była niewystarczająca do indukcji transformacji melanocytów. Wyniki te wskazują, że hipoksyjne środowisko może być promotorem onkogennych właściwości białka c-Kit w melanocytach, a pośrednikiem tego procesu jest czynnik HIF-1α.
O znaczeniu HIF-1 w dalszym rozwoju czerniaka świadczy jego konstytutywna ekspresja w komórkach czerniaka [71] oraz jej wzrost towarzyszący progresji nowotworu [87]. Komórki wyprowadzone ze zmiany nowotworowej w fazie VGP i przerzutów cechują się większą ekspresją tego czynnika w porównaniu z melanocytami i liniami wyizolowanymi ze zmian w fazie RGP [68,87]. Oprócz niedotlenowania i zmian genetycznych również działanie promieniowania UV oraz metali ciężkich pozytywnie reguluje ekspresję tego czynnika w melanocytach [97]. Autorzy pracy szczegółowo opisują rolę czynnika HIF-1 w transformacji nowotworowej nie tylko melanocytów, ale również keratynocytów. Co ciekawe HIF-1 jest mediatorem śmierci apoptotycznej keratynocytów pod wpływem działania promieni UVB [94].
Podsumowując, ekspresja czynnika HIF-1 w melanocytach/komórkach czerniaka może być regulowana różnymi czynnikami. Począwszy od niedotlenowania, przez zmiany genetyczne, po działanie czynników makrośrodowska, np. UV i metale ciężkie. Wszystkie one przyczyniają się do stałej aktywacji ścieżki HIF-1.
Onkogenna aktywność HIF-1/hipoksji w melanocytach wiąże się najprawdopodobniej z regulacją istotnych w rozwoju czerniaka ścieżek oraz ich białek efektorowych. Jednym z nich jest wpływ HIF-1 na aktywność p53. Sendoel i wsp. [121] na modelu Caenorhabditis elegans wykazali, że HIF-1 hamuje proapoptotyczną aktywność białka p53 poprzez indukcje ekspresji białka z rodziny tyrozyn, TYR-2. Białko to jest antagonistą apoptozy indukowanej przez p53. Brak jego aktywności w komórkach czerniaka prowadzi do wzrostu intensywności apoptozy. Niewykluczone, że hamowanie proapoptotycznej aktywności p53, mediowane przez czynnik HIF-1, spełnia warunek konieczny pełnej transformacji melanocytów z mutacją onkogenu BRAF lub aktywną ścieżkę AKT, czyli zablokowanie ścieżek sygnałowych regulujących cykl komórkowy i/lub śmierć komórki.
HIF-1 zmniejsza również ekspresję kluczowego w transformacji melanocytów białka adhezyjnego, E-kadheryny, przez aktywację inhibitora jego ekspresji, białka SNAIL. Pośrednikiem w tej molekularnej kaskadzie jest białko LOX, którego ekspresja znajduje się pod kontrolą czynnika HIF-1 i wzrasta w warunkach niedotlenowania [78]. E-kadheryna jest również efektorem białka NOTCH-1. Aktywacja ścieżki Notch-1 w początkowych etapach transformacji nowotworowej powoduje obniżenie ekspresji E-kadheryny i zwiększenie ekspresji innego ważnego w progresji czerniaka białka adhezyjnego, MCAM [101]. Ścieżka NOTCH jest ściśle powiązana z hipoksyjnym mikrośrodowiskiem, gdyż białko NOTCH-1 jest pod bezpośrednią regulacją właśnie czynnika HIF-1, jak również ścieżki Akt [8].
Niewykluczone, że warunki hipoksji oraz czynnik HIF-1 dodatkowo promują przeżycie i proliferację melanocytów w wyniku aktywacji dwóch najistotniejszych ścieżek sygnałowych w czerniaku, MAPK i PI3K [69,134], skądinąd prowadzących do wzrostu ekspresji samego czynnika HIF-1. Interakcja tych dwóch szlaków może prowadzić do dodatniego sprzężenia zwrotnego napędzającego proliferację i wzrost komórek. Regulację aktywności czynnika HIF-1 oraz jego wybrane cele molekularne przedstawia rycina 5.

Ryc. 5. Regulacja aktywności czynnika HIF-1 w komórkach czerniaka. Podstawowy czynnik transkrypcyjny regulujący odpowiedź na hipoksję może być aktywowany w wyniku działania zarówno czynników zewnętrznych, takich jak niskie stężenie tlenu, promieniowanie UV oraz metale ciężki, jak również w wyniku aktywacji ścieżek sygnałowych związanych z BRAF, MITF, czy PTEN. Efektem jego aktywności jest indukcja ekspresji białek kluczowych w rozwoju czerniaka (np. NOTCH-1, LOX, VEGF) oraz represja supresorów np. p53, CDH-1
Hipoksja może również promować rozwój czerniaka przez upośledzenie odpowiedzi immunologicznej. Noman i wsp.[93] wykazali, że w warunkach niedotlenowania komórki mysiego czerniaka są bardziej oporne na lizę wywołaną działaniem komórek T cytotoksycznych. Jest to spowodowane zwiększoną intensywnością procesu autofagii w komórkach czerniaka pod wpływem hipoksji.
Podsumowując, powyższe wyniki świadczą, że hipoksja w pewnym kontekście molekularnym, np. w przypadku występowania mutacji w genach BRAF, RAS lub c-KIT, może prowadzić do nabycia przez komórki zdolności wzrostu niezależnego od zewnętrznych czynników wzrostu oraz od kontroli keratynocytów, a także zdolności przeciwdziałania procesowi starzenia. Są to warunki niezbędne do przejścia z etapu znamienia barwnikowego do zmiany nowotworowej (RGP). Intensywnie proliferujące komórki są jednocześnie oporne na działanie limfocytów T, co może powodować szybki wzrost nowotworu, zaś pogłębiający się stan hipoksji prowadzi do nabycia przez komórki czerniaka inwazyjnego fenotypu [39].
Hipoksja a inwazyjny fenotyp czerniaka i tworzenie przerzutów
Czerniak w fazie wzrostu wertykalnego cechuje się zdolnością inwazji okolicznych tkanek i tworzenia przerzutów. Hipoksja oraz główne czynniki biorące udział w odpowiedzi na niedotlenowanie mają udokumentowany wpływ na proces inwazji i przerzutowania w wielu typach nowotworów. Obniżone ciśnienie tlenu indukuje zmiany w komórkach nowotworowych prowadzące do selekcji agresywnego, sprzyjającego inwazji fenotypu oraz tworzenia przerzutów. Wykazano, że hipoksja promuje każdy etap progresji nowotworu – od procesu transformacji mezenchymalno-epitelialnej komórek, przez etap intrawazacji, przeżycie komórek w krwiobiegu i ekstrawazację, po wzrost w miejscu utworzenia przerzutu [83].
Hipoksja jest jednym z czynników mikrośrodowiska nowotworowego, który może promować inwazyjny fenotyp również komórek czerniaka. Już w latach 90 ub.w. Stackpol i wsp. [128] wykazali, że komórki mysiego czerniaka poddane działaniu hipoksji intensywniej proliferują, są bardziej inwazyjne oraz łatwiej tworzą przerzuty. Podobne obserwacje poczyniono dla komórek ludzkiego czerniaka oka (Mum2B), które w warunkach hipoksji wykazywały większą zdolność migracji, inwazji i adhezji [138]. Zmiany te były zależne od czynnika HIF-1 i towarzyszyła im ekspresja m.in. genu kodującego receptor chemokiny 4, CXCR4, którego wpływ na inwazyjność i przerzutowanie komórek różnych typów nowotworów jest dobrze udokumentowany [131]. Wzrost agresywności komórek czerniaka pod wpływem hipoksji i ograniczonych zasobów składników odżywczych obserwowali również Osawa i wsp. [97]. Komórki mysiego czerniaka poddane działaniu podwójnego stresu wykazywały zwiększony potencjał proliferacyjny in vitro oraz wzrost tumorogenności i inwazyjności in vivo, najprawdopodobniej związany z aktywacją ścieżki Akt lub nadekspresją czynnika Fgf21.
Hipoksyjne mikrośrodowisko sprzyja również procesowi przerzutowania komórek czerniaka. Obserwowano większą, w porównaniu z grupą zwierząt kontrolnych, liczbę guzów w płucach myszy, której wstrzykiwano dożylnie komórki czerniaka B16 uprzednio poddane działaniu obniżonego stężenia tlenu [104]. Wykazano również dodatnią korelację między liczbą spontanicznych przerzutów ludzkich czerniaków (D-12, R-18) przeszczepionych myszom a wielkością obszarów hipoksji w ich guzach pierwotnych. W badaniu tym guzy bardziej hipoksyjne wykazywały większy potencjał przerzutowania [112]. Dalsze badania potwierdziły wpływ hipoksji zarówno na unaczynienie jak i przerzutowanie [113], aczkolwiek kolejne analizy wykazały, że w większym stopniu niż stan oksygenacji guza rozsiewowi komórek sprzyja potencjał angiogenny komórek i stopień unaczynienia guza [114]. Te pozornie rozbieżne wyniki mogą sugerować, że istotniejszy w rozwoju nowotworu jest hipoksyjny fenotyp jego komórek niż stan hipoksji guza. Przypuszczenia te potwierdzają badania Qi i wsp. [107]. Autorzy analizowali wpływ zahamowania białka Siah2 na wzrost i progresję czerniaka. Siah2 jest ligazą ubikwityny, która reguluje m.in. degradację hydroksylazy prolinowej 3 (Phd3), uczestniczącej w stabilizacji podjednostki alfa HIF-1. Zahamowanie aktywności Siah2 prowadziło do degradacji HIF-1α w komórkach czerniaka i znaczącej redukcji przerzutów tego nowotworu do płuc myszy. Powstałe, nieliczne przerzuty w porównaniu ze zmianami w grupie zwierząt kontrolnych cechował zmniejszony potencjał proliferacyjny, niskie unaczynienie oraz intensywna apoptoza.
W komórkach nowotworowych aktywność HIF-1 jest wynikiem niedotlenowania lub czynników niezależnych od poziomu tlenu, np. aktywacji onkogenów, zahamowania supresorów lub działania niektórych czynników wzrostu i cytokin [10,67,120]. W czerniaku odpowiedź na naturalną hipoksję mikrośrodowiska skóry może być potęgowana aktywnością onkogenu BRAF na wczesnym etapie powstawania czerniaka, a onkogenu MITF na dalszym etapie rozwoju tego nowotworu. Oba te białka stymulują ekspresję czynnika HIF-1α w komórkach czerniaka i melanocytach, o czym wspomniano wcześniej. Niewykluczone, że gdy na początkowym etapie melanomagenezy istotna jest zarówno aktywacja prożyciowej ścieżki HIF-1, jak również pogłębiające się w miarę wzrostu nowotworu niedotlenowanie, będące swoistym czynnikiem selekcjonującym lepiej przystosowane i bardziej agresywne komórki [129], to w dalszym etapie progresji znaczenie selekcji maleje, natomiast istotna staje się indukcja agresywnego fenotypu hipoksyjnego poprzez bezpośrednią regulację ekspresji genów [60] lub zmiany epigenetyczne [123].
Zdolność rozsiewu komórek nowotworowych jest ściśle powiązana z obecnością naczyń krwionośnych i limfatycznych w guzie nowotworowym. Nie inaczej jest w czerniaku. Stan niedotlenowania jest najważniejszym czynnikiem indukującym proces tworzenia nowych naczyń krwionośnych, czyli angiogenezy. Pod kontrolą głównego regulatora odpowiedzi na hipoksję, czynnika HIF-1, znajduje się białka proangiogenne, w tym najważniejszy stymulator angiogenezy, VEGF [132]. Wykazano, że hipoksja powoduje wzrost ekspresji VEGF w komórkach czerniaka z aktywnym białkiem BCL-2 [134]. Jego ekspresję wykrywano również w zmianach czerniakowych oraz znamionach barwnikowych [102]. Ta ostatnia obserwacja potwierdza, że ścieżka HIF-1 jest aktywna już na wczesnych etapach rozwoju czerniaka.
Hipoksja może mieć również wpływ na progresję nowotworu poprzez promowanie tworzenia alternatywnych dróg rozsiewu komórek, czyli mimikrę naczyniową. Sun i wsp. [130] obserwowali zwiększoną liczbę kanałów imitujących naczynia w niedotlenowanych guzach mysiego czerniaka B16. Na tym samym modelu, grupa chińska [145] wykazała, że proces ten może być stymulowany przez indukowaną w warunkach hipoksji tzw. „liniową programowaną śmierć nekrotyczną” (lineary patterned programmed cell necrosis, LPPCN). Badacze sugerują, że powstała w wyniku eliminacji komórek wolna przestrzeń w mikrośrodowisku nowotworowym może być wykorzystywana przez komórki uczestniczące w mimikrze naczyniowej.
O wpływie obniżonego stężenia tlenu w mikrośrodowisku na progresję czerniaka pośrednio świadczy korelacja między ekspresją genów odpowiedzi na hipoksję a przeżyciem pacjentów. Oprócz wspomnianego wyżej VEGF, Giatromonolaki i wsp. [36] wykazali taką zależność dla HIF-2, którego ekspresję obserwowano w około 60% przypadków analizowanych czerniaków, a podwyższony poziom tego białka wiązał się z krótszym przeżyciem pacjentów. Dla innego genu regulowanego hipoksją, galektyny 3, wykazano zwiększoną ekspresję w czerniaku pierwotnym i przerzutach w porównaniu z ekspresją w znamionach barwnikowych [105].
Korelację ekspresji genów aktywowanych czynnikiem HIF-1, takich jak MMP, VEGFA czy COX-2, z inwazyjnością komórek czerniaka wykazali również Spinella i wsp. [127]. Na związek hipoksji z progresją czerniaka pośrednio może wskazywać zaangażowanie genów/białek regulowanych obniżonym stężeniem tlenu w procesy warunkujące jego wzrost i progresję, takie jak inwazyjność, transformacja epitelialno-mezenchymalna czy oporność na chemioterapeutyki. Geny, których ekspresja podlega regulacji w warunkach hipoksji wchodzą również w skład wielogenowych klasyfikatorów czerniaka, wyselekcjonowanych techniką mikromacierzy DNA. Zestawienie genów regulowanych hipoksją, których ekspresja wpływa na biologię czerniaka zawiera tabela 1. Schematyczne przedstawienie wpływu hipoksji na rozwój czerniaka przedstawia rycina 6.
Tabela 1. Geny/białka regulowane hipoksją, które mają udokumentowany związek z biologią czerniaka


Ryc. 6. Wpływ hipoksji na powstanie i rozwój czerniaka. Stan niedotlenowania jest nieodłącznym elementem mikrośrodowiska skóry i obok metali ciężkich oraz promieniowania UV jest głównym induktorem czynnika HIF-1 w melanocytach. W kooperacji z aktywnymi ścieżkami MAPK, AKT, czy c-KIT może promować proces transformacji melanocytów. Jednocześnie główne onkogeny czerniaka BRAF i MITF dodatkowo stymulują jego aktywność, natomiast sam czynnik HIF-1 wpływa na aktywność wielu genów kluczowych w rozwoju i progresji czerniaka, która prowadzi do selekcji komórek bardziej agresywnych, łatwiej przerzutujących
Ekspresja genów promujących inwazyjny wzrost komórek czerniaka w warunkach hipoksji jest podstawą nowego modelu rozwoju tego nowotworu zaproponowanego przez Hoek i wsp. [49–51]. Autorzy sugerują, że komórki czerniaka podczas wzrostu przybierają dwa podstawowe fenotypy. Pierwszy, charakteryzuje się intensywną proliferacją i jest regulowany ekspresją czynnika MITF, drugi zaś cechuje się większą inwazyjnością oraz aktywacją ścieżki TGF-β i zahamowaniem ścieżki Wnt. Te dwa stany funkcjonalne mogą występować naprzemiennie, w zależności od warunków panujących w mikrośrodowisku guza. Jednym z czynników promujących fenotyp inwazyjny, zdaniem autorów, jest niedotlenowanie komórek nowotworowych.
Podsumowując, stan obniżonego stężenia tlenu w mikrośrodowisku od początku towarzyszący rozwojowi czerniaka odgrywa istotną rolę na każdym etapie jego progresji. Promuje transformację nowotworową melanocytów, indukuje inwazyjny fenotyp czerniaka i sprzyja procesowi rozsiewu. Głównym regulatorem tych procesów jest czynnik HIF-1 oraz inne białka indukowane w odpowiedzi na hipoksję. Razem z białkami regulującymi interakcje komórek czerniaka z pozostałymi komórkami mikrośrodowiska nowotworowego stanowią one potencjalne markery agresywności rozwijającego się nowotworu, a być może również cele terapeutyczne dla uzupełniającej terapii czerniaka skóry.
Podziękowania
Dziękuję dr Dorocie Butkiewicz oraz dr. Aleksandrowi Sochanikowi za cenne uwagi podczas edytowania niniejszej pracy.
PIŚMIENNICTWO
[1] Akgül B., García-Escudero R., Ekechi C., Steger G., Navsaria H., Pfister H., Storey A.: The E2 protein of human papillomavirus type 8 increases the expression of matrix metalloproteinase-9 in human keratinocytes and organotypic skin cultures. Med. Microbiol. Immunol., 2011; 200: 127-135
[PubMed]
[2] Alonso S.R., Tracey L., Ortiz P., Pérez-Gómez B., Palacios J., Pollán M., Linares J., Serrano S., Sáez-Castillo A.I., Sánchez L., Pajares R., Sánchez-Aguilera A., Artiga M.J., Piris M.A., Rodríguez-Peralto J.L.: A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res., 2007; 67: 3450-3460
[PubMed] [Full Text HTML] [Full Text PDF]
[3] Ariyanayagam-Baksh S.M., Baksh F.K., Swalsky P.A., Finkelstein S.D.: Loss of heterozygosity in the MXI1 gene is a frequent occurrence in melanoma. Mod. Pathol., 2003; 16: 992-995
[PubMed] [Full Text HTML] [Full Text PDF]
[4] Balsamo M., Scordamaglia F., Pietra G., Manzini C., Cantoni C., Boitano M., Queirolo P., Vermi W., Facchetti F., Moretta A., Moretta L., Mingari M.C., Vitale M.: Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA, 2009; 106: 20847-20852
[PubMed] [Full Text HTML] [Full Text PDF]
[5] Basu B., Biswas S., Wrigley J., Sirohi B., Corrie P.: Angiogenesis in cutaneous malignant melanoma and potential therapeutic strategies. Expert. Rev. Anticancer Ther., 2009; 9: 1583-1598
[PubMed]
[6] Bedogni B., Powell M.B.: Hypoxia, melanocytes and melanoma – survival and tumor development in the permissive microenvironment of the skin. Pigment Cell Melanoma Res., 2009; 22: 166-174
[PubMed] [Full Text HTML] [Full Text PDF]
[7] Bedogni B., Warneke J.A., Nickoloff B.J., Giaccia A.J., Powell M.B.: Notch1 is an effector of Akt and hypoxia in melanoma development. J. Clin. Invest., 2008; 118: 3660-3670
[PubMed] [Full Text HTML] [Full Text PDF]
[8] Bedogni B., Welford S.M., Cassarino D.S., Nickoloff B.J., Giaccia A.J., Powell M.B.: The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell, 2005; 8: 443-454
[PubMed] [Full Text HTML] [Full Text PDF]
[9] Belleudi F., Cardinali G., Kovacs D., Picardo M., Torrisi M.R.: KGF promotes paracrine activation of the SCF/c-KIT axis from human keratinocytes to melanoma cells. Transl. Oncol., 2010; 3: 80-90
[PubMed] [Full Text HTML] [Full Text PDF]
[10] Bertout J.A., Patel S.A., Simon M.C.: The impact of O2 availability on human cancer. Nat. Rev. Cancer, 2008; 8: 967-975
[PubMed] [Full Text HTML] [Full Text PDF]
[11] Bianchini F., Massi D., Marconi C., Franchi A., Baroni G., Santucci M., Mannini A., Mugnai G., Calorini L.: Expression of cyclo-oxygenase-2 in macrophages associated with cutaneous melanoma at different stages of progression. Prostaglandins Other Lipid Mediat., 2007; 83: 320-328
[PubMed]
[12] Bierie B., Moses H.L.: Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer, 2006; 6, 506-520
[PubMed]
[13] Bittner M., Meltzer P., Chen Y., Jiang Y., Seftor E., Hendrix M., Radmacher M., Simon R., Yakhini Z., Ben-Dor A., Sampas N., Dougherty E., Wang E., Marincola F., Gooden C., Lueders J., Glatfelter A., Pollock P., Carpten J., Gillanders E., Leja D., Dietrich K., Beaudry C., Berens M., Alberts D., Sondak V.: Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature, 2000; 406: 536-540
[PubMed] [Full Text HTML] [Full Text PDF]
[14] Boone B., Blokx W., De Bacquer D., Lambert J., Ruiter D., Brochez L.: The role of VEGF-C staining in predicting regional metastasis in melanoma. Virchows Arch., 2008; 453: 257-265
[PubMed]
[15] Bronkhorst I.H., Ly L.V., Jordanova E.S., Vrolijk J., Versluis M., Luyten G.P., Jager M.J.: Detection of M2-macrophages in uveal melanoma and relation with survival. Invest. Ophthalmol. Vis Sci., 2011; 52: 643-650
[PubMed] [Full Text HTML] [Full Text PDF]
[16] Brown E.R., Doig T., Anderson N., Brenn T., Doherty V., Xu Y., Bartlett J.M., Smyth J.F., Melton D.W.: Association of galectin-3 expression with melanoma progression and prognosis. Eur. J. Cancer, 2012; 48: 865-874
[PubMed]
[17] Buback F., Renkl A.C., Schulz G., Weiss J.M.: Osteopontin and the skin: multiple emerging roles in cutaneous biology and pathology. Exp. Dermatol., 2009; 18: 750-759
[PubMed]
[18] Busca R., Berra E., Gaggioli C., Khaled M., Bille K., Marchetti B., Thyss R., Fitsialos G., Larribere L., Bertolotto C., Virolle T., Barbry P., Pouysségur J., Ponzio G., Ballotti R.: Hypoxia-inducible factor 1α is a new target of microphthalmia-associated transcription factor (MITF) in melanoma cells. J. Cell Biol., 2005; 170: 49-59
[PubMed] [Full Text HTML] [Full Text PDF]
[19] Caldarola G., Battista C., Pellicano R.: Melanoma onset after estrogen, thyroid, and growth hormone replacement therapy. Clin. Ther., 2010; 32: 57-59
[PubMed]
[20] Cangara H.M., Ishida T., Hara T., Sun L., Toh R., Rikitake Y., Kundu R.K., Quertermous T., Hirata K., Hayashi Y.: Role of endothelial cell-selective adhesion molecule in hematogeneous metastasis. Microvasc. Res., 2010; 80: 133-141
[PubMed]
[21] Chen W., Ma T., Shen X.N., Xia X.F., Xu G.D., Bai X.L., Liang T.B.: Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res., 2012; 72: 1363-1372
[PubMed] [Full Text HTML] [Full Text PDF]
[22] Cheung M., Sharma A., Madhunapantula S.V., Robertson G.P.: Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res., 2008; 68: 3429-3439
[PubMed] [Full Text HTML] [Full Text PDF]
[23] Ch’ng S., Tan S.T.: Genetics, cellular biology and tumor microenvironment of melanoma. Front. Biosci., 2009; 14: 918-928
[PubMed]
[24] Cipponi A., Wieers G., van Baren N., Coulie P.G.: Tumor-infiltrating lymphocytes: apparently good for melanoma patients. But why? Cancer Immunol. Immunother.: 2011; 60: 1153-1160
[PubMed]
[25] Clark W.H. Jr., Elder D.E., Guerry D.4th., Epstein M.N., Greene M.H., Van Horn M.: A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum. Pathol., 1984; 15: 1147-1165
[PubMed]
[26] Cornil I., Theodorescu D., Man S., Herlyn M., Jambrosic J., Kerbel R.S.: Fibroblast cell interactions with human melanoma cells affect tumor cell growth as a function of tumor progression. Proc. Natl. Acad. Sci. USA, 1991; 88: 6028-6032
[PubMed] [Full Text HTML] [Full Text PDF]
[27] Corrie P.G., Basu B., Zaki K.A.: Targeting angiogenesis in melanoma: prospects for the future.Ther. Adv. Med. Oncol., 2010; 2: 367-380
[PubMed] [Full Text HTML] [Full Text PDF]
[28] Cranfield F.D., Douglas C.M.: Young, pregnant and dying – how can we provide the “right” care? Med. J. Aust.: 2009; 191: 616-617
[PubMed]
[29] Daurkin I., Eruslanov E., Stoffs T., Perrin G.Q., Algood C., Gilbert S.M., Rosser C.J., Su L.M., Vieweg J., Kusmartsev S.: Tumor-associated macrophages mediate immunosuppression in the renal cancer microenvironment by activating the 15-lipoxygenase-2 pathway. Cancer Res., 2011; 71: 6400-6409
[PubMed] [Full Text HTML] [Full Text PDF]
[30] Defresne F., Bouzin C., Grandjean M., Dieu M., Raes M., Hatzopoulos A.K., Kupatt C., Feron O.: Preconditioned endothelial progenitor cells reduce formation of melanoma metastases through SPARC-driven cell-cell interactions and endocytosis. Cancer Res., 2011; 71: 4748-4757
[PubMed] [Full Text HTML] [Full Text PDF]
[31] Dewhirst M.W.: Relationships between cycling hypoxia, HIF-1, angiogenesis and oxidative stress. Radiat. Res., 2009; 172: 653-665
[PubMed] [Full Text HTML] [Full Text PDF]
[32] Evans S.M., Schrlau A.E., Chalian A.A., Zhang P., Koch C.J.: Oxygen levels in normal and previously irradiated human skin as assessed by EF5 binding. J. Invest. Dermatol., 2006; 126: 2596-2606
[PubMed] [Full Text HTML] [Full Text PDF]
[33] Facciabene A., Peng X., Hagemann I.S., Balint K., Barchetti A., Wang L.P., Gimotty P.A., Gilks C.B., Lal P., Zhang L., Coukos G.: Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature, 2011; 475: 226-230
[PubMed]
[34] Fröhlich E.: Proteases in cutaneous malignant melanoma: relevance as biomarker and therapeutic target. Cell Mol. Life Sci., 2010; 67: 3947-3960
[PubMed]
[35] Ghislin S., Obino D., Middendorp S., Boggetto N., Alcaide-Loridan C., Deshayes F.: Junctional adhesion molecules are required for melanoma cell lines transendothelial migration in vitro Pigment Cell Melanoma Res., 2011; 24: 504-511
[PubMed]
[36] Giatromanolaki A., Sivridis E., Kouskoukis C., Gatter K.C., Harris A.L., Koukourakis M.I.: Hypoxia-inducible factors 1alpha and 2alpha are related to vascular endothelial growth factor expression and a poorer prognosis in nodular malignant melanomas of the skin. Melanoma Res., 2003; 13: 493-501
[PubMed]
[37] Goetz J.G., Minguet S., Navarro-Lérida I., Lazcano J.J., Samaniego R., Calvo E., Tello M., Osteso-Ibánez T., Pellinen T., Echarri A., Cerezo A., Klein-Szanto A.J., Garcia R., Keely P.J., Sánchez-Mateos P., Cukierman E., Del Pozo M.A.: Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell, 2011; 146: 148-163
[PubMed] [Full Text HTML] [Full Text PDF]
[38] Gupta A., Driscoll M.S.: Do hormones influence melanoma? Facts and controversies. Clin. Dermatol., 2010; 28: 287-292
[PubMed]
[39] Haass N.K., Herlyn M.: Normal human melanocyte homeostasis as a paradigm for understanding melanoma. J. Investig. Dermatol. Symp. Proc., 2005; 10: 153-163
[PubMed]
[40] Handerson T., Berger A., Harigopol M., Rimm D., Nishigori C., Ueda M., Miyoshi E., Taniguchi N., Pawelek J.: Melanophages reside in hypermelanotic, aberrantly glycosylated tumor areas and predict improved outcome in primary cutaneous malignant melanoma. J. Cutan. Pathol., 2007; 34: 679-686
[PubMed]
[41] Harlin H., Meng Y., Peterson A.C., Zha Y., Tretiakova M., Slingluff C., McKee M., Gajewski T.F.: Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res., 2009; 69: 3077-3085
[PubMed] [Full Text HTML] [Full Text PDF]
[42] Hartmann P., Mirtolouei R., Untersberger S., Ziegler W., Hermann Z., Richtig E., Hofmann-Wellenhof R., Grinschgl S., Kerl H., Smolle J.: Non-invasive imaging of tissue PO2 in malignant melanoma of the skin. Melanoma Res., 2006; 16: 479-486
[PubMed]
[43] Hasumi Y., Kłosowska-Wardega A., Furuhashi M., Ostman A., Heldin C.H., Hellberg C.: Identification of a subset of pericytes that respond to combination therapy targeting PDGF and VEGF signaling. Int. J. Cancer, 2007; 121: 2606-2614
[PubMed]
[44] Hegde U.P., Chakraborty N., Kerr P., Grant-Kels J.M.: Melanoma in the elderly patient: relevance of the aging immune system. Clin. Dermatol., 2009; 27: 537-544
[PubMed]
[45] Helfrich I., Edler L., Sucker A., Thomas M., Christian S., Schadendorf D., Augustin H.G.: Angiopoietin-2 levels are associated with disease progression in metastatic malignant melanoma. Clin. Cancer Res., 2009; 15: 1384-1392
[PubMed] [Full Text HTML] [Full Text PDF]
[46] Helfrich I., Schadendorf D.: Blood vessel maturation, vascular phenotype and angiogenic potential in malignant melanoma: one step forward for overcoming anti-angiogenic drug resistance? Mol. Oncol.: 2011; 5: 137-149
[PubMed]
[47] Hendrix M.J., Seftor E.A., Hess A.R., Seftor R.E.: Molecular plasticity of human melanoma cells. Oncogene, 2003; 22: 3070-3075
[PubMed]
[48] Héroult M., Schaffner F., Pfaff D., Prahst C., Kirmse R., Kutschera S., Riedel M., Ludwig T., Vajkoczy P., Graeser R., Augustin H.G.: EphB4 promotes site-specific metastatic tumor cell dissemination by interacting with endothelial cell-expressed ephrinB2. Mol. Cancer Res., 2010; 8: 1297-1309
[PubMed] [Full Text HTML] [Full Text PDF]
[49] Hoek K.S., Eichhoff O.M., Schlegel N.C., Döbbeling U., Kobert N., Schaerer L., Hemmi S., Dummer R.: In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res., 2008; 68: 650-656
[PubMed] [Full Text HTML] [Full Text PDF]
[50] Hoek K.S., Goding C.R.: Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res., 2010; 23: 746-759
[PubMed]
[51] Hoek K.S., Schlegel N.C., Brafford P., Sucker A., Ugurel S., Kumar R., Weber B.L., Nathanson K.L., Phillips D.J., Herlyn M., Schadendorf D., Dummer R.: Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res., 2006; 19: 290-302
[PubMed]
[52] Hsu M.Y., Meier F.E., Nesbit M., Hsu J.Y., Van Belle P., Elder D.E., Herlyn M.: E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am. J. Pathol., 2000; 56: 1515-1525
[PubMed] [Full Text HTML] [Full Text PDF]
[53] Hsu M.Y., Shih D.T., Meier F.E., Van Belle P., Hsu J.Y., Elder D.E., Buck C.A., Herlyn M.: Adenoviral gene transfer of beta3 integrin subunit induces conversion from radial to vertical growth phase in primary human melanoma. Am. J. Pathol., 1998; 153: 1435-1442
[PubMed] [Full Text HTML] [Full Text PDF]
[54] Huang B., Lei Z., Zhang G.M., Li D., Song C., Li B., Liu Y., Yuan Y., Unkeless J., Xiong H., Feng Z.H.: SCF-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood, 2008; 112: 1269-1279
[PubMed] [Full Text HTML] [Full Text PDF]
[55] Huber M.A., Kraut N., Park J.E., Schubert R.D., Rettig W.J., Peter R.U., Garin-Chesa P. Fibroblast activation protein: differential expression and serine protease activity in reactive stromal fibroblasts of melanocytic skin tumors. J. Invest. Dermatol., 2003; 120: 182-188
[PubMed] [Full Text HTML] [Full Text PDF]
[56] Hyter S., Bajaj G., Liang X., Barbacid M., Ganguli-Indra G., Indra A.K.: Loss of nuclear receptor RXRα in epidermal keratinocytes promotes the formation of Cdk4-activated invasive melanomas. Pigment Cell Melanoma Res., 2010; 23: 635-648
[PubMed] [Full Text HTML] [Full Text PDF]
[57] Jaeger J., Koczan D., Thiesen H.J., Ibrahim S.M., Gross G., Spang R., Kunz M.: Gene expression signatures for tumor progression, tumor subtype, and tumor thickness in laser-microdissected melanoma tissues. Clin. Cancer Res., 2007; 13: 806-815
[PubMed] [Full Text HTML] [Full Text PDF]
[58] Jager M.J., Ly L.V., El Filali M., Madigan M.C.: Macrophages in uveal melanoma and in experimental ocular tumor models: Friends or foes? Prog. Retin. Eye Res., 2011; 30: 129-146
[PubMed]
[59] Jamal S., Schneider R.J.: UV-induction of keratinocyte endothelin-1 downregulates E-cadherin in melanocytes and melanoma cells. Clin. Invest., 2002; 110: 443-452
[PubMed] [Full Text HTML] [Full Text PDF]
[60] Kenneth N.S., Rocha S.: Regulation of gene expression by hypoxia. Biochem. J., 2008; 414: 19-29
[PubMed]
[61] Khosrotehrani K., Nguyen H. S., Prignon A., Avril M.F., Boitier F., Oster M., Mortier L., Richard M.A., Maubec E., Kerob D., Mansard S., Merheb C., Moguelet P., Nassar D., Guégan S., Aractingi S.: Pregnancy promotes melanoma metastasis through enhanced lymphangiogenesis. Am. J. Pathol., 2011; 178: 1870-1880
[PubMed] [Full Text HTML] [Full Text PDF]
[62] Kim M., Koh Y.J., Kim K.E., Koh B.I., Nam D.H., Alitalo K., Kim I., Koh G.Y.: CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer Res., 2010; 70: 10411-10421
[PubMed] [Full Text HTML] [Full Text PDF]
[63] Kippenberger S., Loitsch S., Thaçi D., Müller J., Guschel M., Kaufmann R., Bernd A.: Restoration of E-cadherin sensitizes human melanoma cells for apoptosis. Melanoma Res., 2006, 16: 393-403
[PubMed]
[64] Kirschmann D.A., Seftor E.A., Fong S.F., Nieva D.R., Sullivan C.M., Edwards E.M., Sommer P., Csiszar K., Hendrix M.J.: A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res., 2002; 62: 4478-4483
[PubMed] [Full Text HTML] [Full Text PDF]
[65] Kluk M.J., Grant-Kels J.M., Kerr P., Hoss D., Berke A., Claffey K.P., Murphy M.: Melanoma on the move: the progression of melanoma: novel concepts with histologic correlates. Am. J. Dermatopathol., 2004; 26: 504-510
[PubMed]
[66] Koefinger P., Wels C., Joshi S., Damm S., Steinbauer E., Beham-Schmid C., Frank S., Bergler H., Schaider H.: The cadherin switch in melanoma instigated by HGF is mediated through epithelial-mesenchymal transition regulators. Pigment Cell Melanoma Res., 2011; 24: 382-385
[PubMed] [Full Text HTML] [Full Text PDF]
[67] Koh M.Y., Darnay B.G., Powis G.: Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol. Cell Biol., 2008; 28: 7081-7095
[PubMed] [Full Text HTML] [Full Text PDF]
[68] Kumar S.M., Yu H., Edwards R., Chen L., Kazianis S., Brafford P., Acs G., Herlyn M., Xu X.: Mutant V600E BRAF increases hypoxia inducible factor-1alpha expression in melanoma. Cancer Res., 2007; 67: 3177-3184
[PubMed] [Full Text HTML] [Full Text PDF]
[69] Kunz M., Ibrahim S., Koczan D., Thiesen H.J., Köhler H.J., Acker T., Plate K.H., Ludwig S., Rapp U.R., Bröcker E.B., van Muijen G.N., Flory E., Gross G.: Activation of c-Jun NH2-terminal kinase/stress-activated protein kinase (JNK/SAPK) is critical for hypoxia-induced apoptosis of human malignant melanoma. Cell Growth Differ., 2001; 12: 137-145
[PubMed] [Full Text HTML] [Full Text PDF]
[70] Kunz M., Moeller S., Koczan D., Lorenz P., Wenger R.H., Glocker M.O., Thiesen H.J., Gross G., Ibrahim S.M.: Mechanisms of hypoxic gene regulation of angiogenesis factor Cyr61 in melanoma cells. J. Biol. Chem., 2003; 278: 45651-45660
[PubMed] [Full Text HTML] [Full Text PDF]
[71] Kuphal S., Winklmeier A., Warnecke C., Bosserhoff A.K.: Constitutive HIF-1 activity in malignant melanoma. Eur. J. Cancer, 2010; 46: 1159-1169
[PubMed]
[72] Labrousse A.L., Ntayi C., Hornebeck W., Bernard P.: Stromal reaction in cutaneous melanoma. Crit. Rev. Oncol. Hematol., 2004; 49: 269-275
[PubMed]
[73] Ladányi A., Kiss J., Mohos A., Somlai B., Liszkay G., Gilde K., Fejős Z., Gaudi I., Dobos J., Tímár J.: Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol. Immunother., 2011; 60: 1729-1738
[PubMed]
[74] Langer H.F., Orlova V.V., Xie C., Kaul S., Schneider D., Lonsdorf A.S., Fahrleitner M., Choi E.Y., Dutoit V., Pellegrini M., Grossklaus S., Nawroth P.P., Baretton G., Santoso S., Hwang S.T., Arnold B., Chavakis T.: A novel function of junctional adhesion molecule-C in mediating melanoma cell metastasis. Cancer Res., 2011; 71: 4096-4105
[PubMed] [Full Text HTML] [Full Text PDF]
[75] Lartigau E., Randrianarivelo H., Avril M.F., Margulis A., Spatz A., Eschwege F., Guichard M.: Intratumoral oxygen tension in metastatic melanoma. Melanoma Res., 1997; 7: 400-406
[PubMed]
[76] Lazova R., Chakraborty A., Pawelek J.M.: Leukocyte-cancer cell fusion: initiator of the warburg effect in malignancy? Adv. Exp. Med. Biol., 2011; 714: 151-172
[PubMed]
[77] Lee J., Fassnacht M., Nair S., Boczkowski D., Gilboa E.: Tumor immunotherapy targeting fibroblast activation protein, a product expressed in tumor-associated fibroblasts. Cancer Res., 2005; 65: 11156-11163
[PubMed] [Full Text HTML] [Full Text PDF]
[78] Lee J.T., Herlyn M.: Microenvironmental influences in melanoma progression. J. Cell Biochem., 2007; 101: 862-872
[PubMed]
[79] Li G., Fukunaga M., Herlyn M.: Reversal of melanocytic malignancy by keratinocytes is an E-cadherin-mediated process overriding β-catenin signaling. Exp. Cell Res., 2004; 297: 142-151
[PubMed]
[80] Li G., Satyamoorthy K., Herlyn M.: Dynamics of cell interactions and communications during melanoma development. Crit. Rev. Oral Biol. Med., 2002; 13: 62-70
[PubMed]
[81] Liang S., Hoskins M., Dong C.: Tumor cell extravasation mediated by leukocyte adhesion is shear rate dependent on IL-8 signaling. Mol. Cell Biomech., 2010; 7: 77-91
[PubMed] [Full Text HTML] [Full Text PDF]
[82] Liu B., Ma J., Wang X., Su F., Li X., Yang S., Ma W., Zhang Y.: Lymphangiogenesis and its relationship with lymphatic metastasis and prognosis in malignant melanoma. Anat. Rec. (Hoboken), 2008; 291: 1227-1235
[PubMed] [Full Text HTML] [Full Text PDF]
[83] Lu X., Kang Y.: Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin. Cancer Res., 2010; 16: 5928-5935
[PubMed] [Full Text HTML] [Full Text PDF]
[84] Mahabeleshwar G.H., Byzova T.V.: Angiogenesis in melanoma. Semin. Oncol., 2007; 34: 555-565
[PubMed] [Full Text HTML] [Full Text PDF]
[85] Markovic S.N., Erickson L.A., Rao R.D., Weenig R.H., Pockaj B.A., Bardia A., Vachon C.M., Schild S.E., McWilliams R.R., Hand J.L., Laman S.D., Kottschade L.A., Maples W.J., Pittelkow M.R., Pulido J.S., Cameron J.D., Creagan E.T.: Melanoma Study Group of Mayo Clinic Cancer Center. Malignant melanoma in the 21st century, part 2: staging, prognosis, and treatment.. Mayo Clin. Proc., 2007; 82: 490-513
[PubMed]
[86] Melnikova V.O., Bar-Eli M.: Bioimmunotherapy for melanoma using fully human antibodies targeting MCAM/MUC18 and IL-8. Pigment Cell Res., 2006; 19: 395-405
[PubMed]
[87] Mills C.N., Joshi S.S., Niles R.M.: Expression and function of hypoxia inducible factor-1 alpha in human melanoma under non-hypoxic conditions. Mol. Cancer, 2009; 8: 104
[PubMed] [Full Text HTML] [Full Text PDF]
[88] Monsel G., Ortonne N., Bagot M., Bensussan A., Dumaz N.: c-Kit mutants require hypoxia-inducible factor 1alpha to transform melanocytes. Oncogene, 2010; 29: 227-236
[PubMed]
[89] Mrochem J., Bartnik W.: Osteopontyna – nowy marker w chorobach nowotworowych. Wsp. Onkol., 2008; 12: 349-353
[Full Text HTML]
[90] Müller M., Beck I.M., Gadesmann J., Karschuk N., Paschen A., Proksch E., Djonov V., Reiss K., Sedlacek R.: MMP19 is upregulated during melanoma progression and increases invasion of melanoma cells. Mod. Pathol., 2010; 23: 511-521
[PubMed] [Full Text HTML] [Full Text PDF]
[91] Navarini-Meury A.A., Conrad C.: Melanoma and innate immunity – active inflammation or just erroneous attraction? Melanoma as the source of leukocyte-attracting chemokines. Semin Cancer Biol., 2009; 19: 84-91
[PubMed]
[92] Nisancioglu M.H., Betsholtz C., Genové G.: The absence of pericytes does not increase the sensitivity of tumor vasculature to vascular endothelial growth factor-A blockade. Cancer Res., 2010; 70: 5109-5115
[PubMed] [Full Text HTML] [Full Text PDF]
[93] Noman M.Z., Janji B., Kaminska B., Van Moer K., Pierson S., Przanowski P., Buart S., Berchem G., Romero P., Mami-Chouaib F., Chouaib S.: Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res., 2011; 71: 5976-5986
[PubMed] [Full Text HTML] [Full Text PDF]
[94] Nys K., Maes H., Dudek A.M., Agostinis P.: Uncovering the role of hypoxia inducible factor-1α in skin carcinogenesis. Biochim. Biophys. Acta, 2011; 1816: 1-12
[PubMed]
[95] Nys K., Van Laethem A., Michiels C., Rubio N., Piette J.G., Garmyn M., Agostinis P.: A p38MAPK/HIF-1 pathway initiated by UVB irradiation is required to induce Noxa and apoptosis of human keratinocytes. J. Invest. Dermatol., 2010; 130: 2269-2276
[PubMed] [Full Text HTML] [Full Text PDF]
[96] Orimo A., Weinberg R.A.: Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle, 2006; 5: 1597-1601
[PubMed] [Full Text PDF]
[97] Osawa T., Muramatsu M., Watanabe M., Shibuya M.: Hypoxia and low-nutrition double stress induces aggressiveness in a murine model of melanoma. Cancer Sci., 2009; 100: 844-851
[PubMed]
[98] Ozerdem U.: Targeting pericytes diminishes neovascularization in orthotopic uveal melanoma in NG2 proteoglycan knockout mouse. Ophthalmic Res., 2006; 38: 251-254
[PubMed] [Full Text HTML] [Full Text PDF]
[99] Panelos J., Massi D.: Emerging role of Notch signaling in epidermal differentiation and skin cancer. Cancer Biol. Ther., 2009; 8: 1986-1993
[PubMed] [Full Text HTML] [Full Text PDF]
[100] Paradela S., Fonseca E., Pita-Fernandez S., Kantrow S.M., Goncharuk V.N., Ivan D., Herzog C.E., Sturgis E.M., Prieto V.G.: Melanoma under 18 years and pregnancy: report of three cases. Eur. J. Dermatol., 2010; 20: 186-188
[PubMed] [Full Text HTML] [Full Text PDF]
[101] Pinnix C.C., Lee J.T., Liu Z.J., McDaid R., Balint K., Beverly L.J., Brafford P.A., Xiao M., Himes B., Zabierowski S.E., Yashiro-Ohtani Y., Nathanson K.L., Bengston A., Pollock P.M., Weeraratna A.T., Nickoloff B.J., Pear W.S., Capobianco A.J., Herlyn M.: Active Notch1 confers a transformed phenotype to primary human melanocytes. Cancer Res., 2009; 69: 5312-5320
[PubMed] [Full Text HTML] [Full Text PDF]
[102] Pisacane A.M., Risio M.: VEGF and VEGFR-2 immunohistochemistry in human melanocytic naevi and cutaneous melanomas. Melanoma Res., 2005; 15: 39-43
[PubMed]
[103] Pollard J.W.: Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer, 2004; 4: 71-78
[PubMed]
[104] Postovit L.M., Adams M.A., Lash G.E., Heaton J.P., Graham C.H.: Nitric oxide-mediated regulation of hypoxia-induced B16F10 melanoma metastasis. Int. J. Cancer, 2004; 108: 47-53
[PubMed]
[105] Prieto V.G., Mourad-Zeidan A.A., Melnikova V., Johnson M.M., Lopez A., Diwan A.H., Lazar A.J., Shen S.S., Zhang P.S., Reed J.A., Gershenwald J.E., Raz A., Bar-Eli M.: Galectin-3 expression is associated with tumor progression and pattern of sun exposure in melanoma. Clin. Cancer Res., 2006; 12: 6709-6715
[PubMed] [Full Text HTML] [Full Text PDF]
[106] Puré E.: The road to integrative cancer therapies: emergence of a tumor-associated fibroblast protease as a potential therapeutic target in cancer. Expert Opin. Ther. Targets, 2009; 13: 967-973
[PubMed]
[107] Qi J., Nakayama K., Gaitonde S., Goydos J.S., Krajewski S., Eroshkin A., Bar-Sagi D., Bowtell D., Ronai Z.: The ubiquitin ligase Siah2 regulates tumorigenesis and metastasis by HIF-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA, 2008; 105: 16713-16718
[PubMed] [Full Text HTML] [Full Text PDF]
[108] Qian B.Z., Pollard J.W.: Macrophage diversity enhances tumor progression and metastasis. Cell, 2010; 141: 39-51
[PubMed] [Full Text HTML] [Full Text PDF]
[109] Ramirez-Montagut T., Blachere N.E., Sviderskaya E.V., Bennett D.C., Rettig W.J., Garin-Chesa P., Houghton A.N.: FAPα, a surface peptidase expressed during wound healing, is a tumor suppressor. Oncogene, 2004; 23: 5435-5446
[PubMed]
[110] Rettig W.J., Garin-Chesa P., Healey J.H., Su S.L., Ozer H.L., Schwab M., Albino A.P., Old L.J.: Regulation and heteromeric structure of the fibroblast activation protein in normal and transformed cells of mesenchymal and neuroectodermal origin. Cancer Res., 1993; 53: 3327-3335
[PubMed] [Full Text HTML] [Full Text PDF]
[111] Roda J.M., Sumner L.A., Evans R., Phillips G.S., Marsh C.B., Eubank T.D.: Hypoxia-inducible factor-2α regulates GM-CSF-derived soluble vascular endothelial growth factor receptor 1 production from macrophages and inhibits tumor growth and angiogenesis. J. Immunol., 2011; 187: 1970-1976
[PubMed] [Full Text HTML] [Full Text PDF]
[112] Rofstad E.K., Galappathi K., Mathiesen B., Ruud E.B.: Fluctuating and diffusion-limited hypoxia in hypoxia-induced metastasis. Clin. Cancer Res., 2007; 13: 1971-1978
[PubMed] [Full Text HTML] [Full Text PDF]
[113] Rofstad E.K., Gaustad J.V., Egeland T.A., Mathiesen B., Galappathi K.: Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int. J. Cancer, 2010; 127: 1535-1546
[PubMed]
[114] Rofstad E.K., Mathiesen B.: Metastasis in melanoma xenografts is associated with tumor microvascular density rather than extent of hypoxia. Neoplasia, 2010; 12: 889-898
[PubMed] [Full Text HTML] [Full Text PDF]
[115] Roy M., Pear W.S., Aster J.C.: The multifaceted role of Notch in cancer. Curr. Opin. Genet. Dev., 2007; 17: 52-59
[PubMed]
[116] Ruiter D., Bogenrieder T., Elder D., Herlyn M.: Melanoma-stroma interactions: structural and functional aspects. Lancet Oncol., 2002; 3: 35-43
[PubMed]
[117] Sacewicz I., Wiktorska M., Wysocki T., Niewiarowska J.: Mechanizmy angiogenezy nowotworowej. Postępy Hig. Med. Dośw., 2009; 63: 159-168
[PubMed] [Full Text HTML] [Full Text PDF]
[118] Sato T., Ishiko A., Saito M., Tanaka M., Ishimoto H., Amagai M.: Rapid growth of malignant melanoma in pregnancy. J. Dtsch. Dermatol. Ges., 2008; 6: 126-129
[PubMed]
[119] Seftor E.A., Brown K.M., Chin L., Kirschmann D.A., Wheaton W.W., Protopopov A., Feng B., Balagurunathan Y., Trent J.M., Nickoloff B.J., Seftor R.E., Hendrix M.J.: Epigenetic transdifferentiation of normal melanocytes by a metastatic melanoma microenvironment. Cancer Res., 2005; 65: 10164-10169
[PubMed] [Full Text HTML] [Full Text PDF]
[120] Semenza G.: Signal transduction to hypoxia-inducible factor 1. Biochem. Pharmacol., 2002; 64: 993-998
[PubMed]
[121] Sendoel A., Kohler I., Fellmann C., Lowe S.W., Hengartner M.O.: HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature, 2010; 465: 577-583
[PubMed] [Full Text HTML] [Full Text PDF]
[122] Sethi G., Shanmugam M.K., Ramachandran L., Kumar A.P., Tergaonkar V.: Multifaceted link between cancer and inflammation. Biosci. Rep., 2012; 32: 1-15
[PubMed] [Full Text HTML] [Full Text PDF]
[123] Shahrzad S., Bertrand K., Minhas K., Coomber B.L.: Induction of DNA hypomethylation by tumor hypoxia. Epigenetics, 2007; 2: 119-125
[PubMed] [Full Text HTML] [Full Text PDF]
[124] Shao H., Cai L., Grichnik J.M., Livingstone A.S., Velazquez O.C., Liu Z.J.: Activation of Notch1 signaling in stromal fibroblasts inhibits melanoma growth by upregulating WISP-1. Oncogene, 2011; 30: 4316-4326
[PubMed]
[125] Shellman Y.G., Makela M., Norris D.A.: Induction of secreted matrix metalloproteinase-9 activity in human melanoma cells by extracellular matrix proteins and cytokines. Melanoma Res., 2006; 16: 207-211
[PubMed]
[126] Shi L., Lei D., Ma C., Xu F., Li Y., Wang Y., Cong N., Liu D., Pan X.L. Clinicopathological implications of tumour-associated macrophages and vascularization in sinonasal melanoma. J. Int. Med. Res., 2010; 38: 1276-1286
[PubMed]
[127] Spinella F., Rosano L., Di Castro V., Decandia S., Nicotra M.R., Natali P.G., Bagnato A.: Endothelin-1 and endothelin-3 promote invasive behavior via hypoxia-inducible factor-1alpha in human melanoma cells. Cancer Res., 2007; 67: 1725-1734
[PubMed] [Full Text HTML] [Full Text PDF]
[128] Stackpole C.W., Groszek L., Kalbag S.S.: Benign-to-malignant B16 melanoma progression induced in two stages in vitro by exposure to hypoxia. J. Natl. Cancer Inst., 1994; 86: 361-367
[PubMed]
[129] Sullivan R., Graham C.H.: Hypoxia-driven selection of the metastatic phenotype. Cancer Metastasis Rev., 2007; 26: 319-331
[PubMed]
[130] Sun B., Zhang D., Zhang S., Zhang W., Guo H., Zhao X.: Hypoxia influences vasculogenic mimicry channel formation and tumor invasion-related protein expression in melanoma. Cancer Lett., 2007; 249: 188-197
[PubMed]
[131] Sun X., Cheng G., Hao M., Zheng J., Zhou X., Zhang J., Taichman R.S., Pienta K.J., Wang J.: CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev., 2010; 29: 709-722
[PubMed] [Full Text HTML] [Full Text PDF]
[132] Szala S., Jarosz M.: Nowotworowe naczynia krwionośne. Postępy Hig. Med. Dośw., 2011; 65: 437-446
[PubMed] [Full Text HTML] [Full Text PDF]
[133] Tang A., Eller M.S., Hara M., Yaar M., Hirohashi S., Gilchrest B.A.: E-cadherin is the major mediator of human melanocyte adhesion to keratinocytes in vitro. J. Cell Sci., 1994; 107: 983-992
[PubMed] [Full Text HTML] [Full Text PDF]
[134] Trisciuoglio D., Iervolino A., Zupi G., Del Bufalo D.: Involvement of PI3K and MAPK signaling in bcl-2-induced vascular endothelial growth factor expression in melanoma cells. Mol. Biol. Cell, 2005; 16: 4153-4162
[PubMed] [Full Text HTML] [Full Text PDF]
[135] Troyanova P.: The role of trauma in the melanoma formation. J. BUON, 2002; 7: 347-350
[PubMed]
[136] Turner F.E., Broad S., Khanim F.L., Jeanes A., Talma S., Hughes S., Tselepis C., Hotchin N.A.: Slug regulates integrin expression and cell proliferation in human epidermal keratinocytes. J. Biol. Chem., 2006; 281: 21321-21331
[PubMed] [Full Text HTML] [Full Text PDF]
[137] Van Kilsdonk J.W., Bergers M., Van Kempen L.C., Schalkwijk J., Swart G.W. Keratinocytes drive melanoma invasion in a reconstructed skin model. Melanoma Res., 2010; 20: 372-380
[PubMed]
[138] Victor N., Ivy A., Jiang B.H., Agani F.H.: Involvement of HIF-1 in invasion of Mum2B uveal melanoma cells. Clin. Exp. Metastasis, 2006; 23: 87-96
[PubMed]
[139] Villanueva J., Herlyn M.: Melanoma and the tumor microenvironment. Curr. Oncol. Rep., 2008; 10: 439-446
[PubMed]
[140] Wandel E., Grasshoff A., Mittag M., Haustein U.F., Saalbach A.: Fibroblasts surrounding melanoma express elevated levels of matrix metalloproteinase-1 (MMP-1) and intercellular adhesion molecule-1 (ICAM-1) in vitro. Exp. Dermatol., 2000; 9: 34-41
[PubMed]
[141] Wang E., Miller L.D., Ohnmacht G.A., Mocellin S., Perez-Diez A., Petersen D., Zhao Y., Simon R., Powell J.I., Asaki E., Alexander H.R., Duray P.H., Herlyn M., Restifo N.P., Liu E.T., Rosenberg S.A., Marincola F.M.: Prospective molecular profiling of melanoma metastases suggests classifiers of immune responsiveness. Cancer Res., 2002; 62: 3581-3586
[PubMed] [Full Text HTML] [Full Text PDF]
[142] Wäster P., Rosdahl I., Gilmore B.F., Seifert O.: Ultraviolet exposure of melanoma cells induces fibroblast activation protein-α in fibroblasts: implications for melanoma invasion. Int. J. Oncol., 2011; 39: 193-202
[PubMed]
[143] Winnepenninckx V., Lazar V., Michiels S., Dessen P., Stas M., Alonso S.R., Avril M.F., Ortiz Romero P.L., Robert T., Balacescu O., Eggermont A.M., Lenoir G., Sarasin A., Tursz T., van den Oord J.J., Spatz A.: Melanoma Group of the European Organization for Research and Treatment of Cancer. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J. Natl. Cancer Inst., 2006; 98: 472-482
[PubMed] [Full Text HTML] [Full Text PDF]
[144] Wu F.H., Yuan Y., Li D., Lei Z., Song C.W., Liu Y.Y., Li B., Huang B., Feng Z.H., Zhang G.M.: Endothelial cell-expressed Tim-3 facilitates metastasis of melanoma cells by activating the NF-κB pathway. Oncol. Rep., 2010; 24: 693-699
[PubMed] [Full Text PDF]
[145] Zhang S., Li M., Zhang D., Xu S., Wang X., Liu Z., Zhao X., Sun B.: Hypoxia influences linearly patterned programmed cell necrosis and tumor blood supply patterns formation in melanoma. Lab. Invest., 2009; 89: 575-586
[PubMed] [Full Text HTML] [Full Text PDF]
[146] Zigler M., Kamiya T., Brantley E.C., Villares G.J., Bar-Eli M.: PAR-1 and thrombin: the ties that bind the microenvironment to melanoma metastasis. Cancer Res.: 2011; 71: 6561-6566
[PubMed] [Full Text HTML] [Full Text PDF]
[147] Zigler M., Villares G.J., Dobroff A.S., Wang H., Huang L., Braeuer R.R., Kamiya T., Melnikova V.O., Song R., Friedman R., Alani R.M., Bar-Eli M.: Expression of Id-1 is regulated by MCAM/MUC18: a missing link in melanoma progression. Cancer Res., 2011; 71: 3494-3504
[PubMed] [Full Text HTML] [Full Text PDF]
[148] Zigrino P., Nischt R., Mauch C.: The disintegrin-like and cysteine-rich domains of ADAM-9 mediate interactions between melanoma cells and fibroblasts. J. Biol. Chem., 2011; 286: 6801-6807
[PubMed] [Full Text HTML] [Full Text PDF]
Autorka deklaruje brak potencjalnych konfliktów interesów.