
The role of glucose transporter 1 (GLUT1) in the diagnosis and therapy of tumors
Paweł Jóźwiak 1 , Anna Lipińska 1Streszczenie
Podstawową cechą komórek nowotworowych jest przyspieszony metabolizm glukozy połączony z zahamowaniem procesu fosforylacji oksydacyjnej. Zwiększone tempo glikolizy umożliwia kompensację niewielkiego zysku energetycznego wynikającego z oddychania beztlenowego, co pozwala komórkom nowotworowym kontynuować procesy niekontrolowanego wzrostu i proliferacji. W nasilonym pobieraniu glukozy przez komórki pośredniczą transportery glukozy określane jako GLUT. Nadekspresję białek GLUT, a zwłaszcza regulowanego warunkami hipoksji GLUT1, opisano w wielu typach nowotworów. Liczne doniesienia wskazują na korelację pomiędzy poziomem ekspresji GLUT1 a stopniem zezłośliwienia nowotworu. Regulacja poziomu GLUT1 stanowi główny czynnik wpływający na metabolizm glukozy w komórkach nowotworowych stając się potencjalnym celem chemioterapii. W pracy przedstawiono obecny stan wiedzy na temat struktury, regulacji oraz znaczenia transportera GLUT1 w diagnostyce i terapii nowotworów.
Słowa kluczowe:GLUT1 • HIF-1 • mTOR • nowotwory • onkogeny • analogi glukozy • glikoliza
Summary
Malignant cells are known to enhance glucose metabolism, to increase glucose uptake and to inhibit the process of oxidative phosphorylation. Accelerated glycolysis is one of the biochemical characteristics of cancer cells that allow them to compensate the inefficient extraction of energy from glucose in order to continue their uncontrolled growth and proliferation. Upregulation of glucose transport across the plasma membrane is mediated by a family of facilitated glucose transporter proteins named GLUT. Overexpression of GLUTs, especially the hypoxia-responsive GLUT1, has been frequently observed in various human carcinomas. Many studies have reported a correlation between GLUT1 expression level and the grade of tumor aggressiveness, which suggests that GLUT1 expression may be of prognostic significance. Therefore, GLUT1 is a key rate-limiting factor in the transport and glucose metabolism in cancer cells. This paper presents the current state of knowledge on GLUT1 regulation as well as its utility in the diagnosis and therapy of cancers.
Key words:GLUT1 • HIF-1 • mTOR • cancer • oncogene • glucose analogs • glycolysis
Wprowadzenie
Powszechnie wiadomo, że komórki nowotworowe charakteryzują się odmiennym metabolizmem w porównaniu z komórkami prawidłowymi. W komórkach prawidłowych w procesie glikolizy glukoza jest metabolizowana do dwóch cząsteczek pirogronianu z wytworzeniem dwóch cząsteczek ATP. Powstały pirogronian w mitochondriach jest utleniany do acetylo-CoA z udziałem kompleksu dehydrogenazy pirogronianowej. Następnie acetylo-CoA wchodzi w cykl Krebsa, w którym ulega utlenieniu do CO2, a zmagazynowana energia w postaci koenzymów NADH i FADH2 podlega przemianie w cząsteczki ATP podczas transportu elektronów poprzez układy oksydoredukcyjne łańcucha oddechowego. W wyniku całkowitego utlenienia cząsteczki glukozy zostaje wytworzone 30 cząsteczek ATP. W komórkach nowotworowych następuje zahamowanie transportu elektronów na tlen oraz procesu fosforylacji oksydacyjnej. To z kolei skutkuje redukcją wytwarzania ATP oraz zwiększeniem generowania reaktywnych form tlenu, prowadzącym do wzmożonych mutacji w obrębie genów [18,21]. W komórkach nowotworowych w warunkach hipoksji jest faworyzowana przemiana pirogronianu do mleczanu poprzez indukcję ekspresji izoformy A dehydrogenazy mleczanowej. W reakcji powstawania mleczanu odtworzony zostaje koenzym NAD+ umożliwiający cykliczność procesu glikolizy, a całkowity zysk energetyczny oddychania beztlenowego stanowią 2 cząsteczki ATP [84,96]. Niewielki zysk energetyczny oddychania beztlenowego w połączeniu z dużymi wymaganiami energetycznymi komórek nowotworowych prowadzi do 20-30-krotnego zintensyfikowania procesu glikolizy oraz zwiększonego pobierania glukozy przez komórkę, które odbywa się z udziałem transporterów glukozy określanych jako GLUT [2,21].
W komórkach człowieka zidentyfikowano 14 białek rodziny GLUT (które zgrupowano w trzy klasy) charakteryzujących się wspólną transmembranową topologią [85]. Białka GLUT transportują heksozy niezależnie od energii, zgodnie z gradientem stężeń, z różną kinetyką i powinowactwem względem substratu. Każda izoforma ma tkankowo swoiste umiejscowienie. Ekspresję więcej niż jednej izoformy można obserwować w tej samej tkance lub wiele tkanek może transportować heksozy z udziałem tego samego (określonego) transportera GLUT w jednym czasie [95].
Transporter glukozy 1 – charakterystyka molekularna
Transporter glukozy 1 (GLUT1) jest najlepiej poznaną i wszechobecnie występującą izoformą rodziny białek GLUT. W stanach fizjologicznych wysoki poziom ekspresji GLUT1 obserwuje się w erytrocytach (3-5% wszystkich białek błonowych), nabłonku endotelialnym i epitelialnym bariery krew-mózg, oku, łożysku i gruczole mlekowym ssaków [98]. Badania swoistości substratowej GLUT1 względem heksoz, ulegającego ekspresji w oocytach Xenopus leavis, wykazały dużą wydajność transportu glukozy (Km~3 mM) [87] i 2-deoksyglukozy (Km~5 mM) [91]. Ponadto GLUT1 może transportować: galaktozę, mannozę, glukozaminę i kwas askorbinowy [34,98].
W komórkach człowieka transporter GLUT1 zbudowany z 492 aminokwasów (m.m. ~54 kDa), jest kodowany przez gen SLC2A1 (złożony z 10 eksonów) umiejscowiony na krótkim ramieniu chromosomu 1 (1p35-31.3). Wszystkie transportery GLUT zawierają 12 hydrofobowych a-heliksowych domen przypuszczalnie 12-krotnie przechodzących przez błonę plazmatyczną. Regiony N- i C-końcowe tych białek oraz duża pętla (zlokalizowana pomiędzy segmentem 6 i 7) znajdują się po stronie cytoplazmatycznej (ryc. 1).

Ryc. 1. Budowa transportera GLUT1 (opis w tekście – wg [26] zmodyfikowano)
Białka klasy I rodziny GLUT, do której należy GLUT1, mają pomiędzy domeną transmembranową 1 i 2 zewnątrzkomórkową pętlę z miejscem glikozylacji [31,98]. N-glikozylacja Asn 45 GLUT1 wpływa na jego stabilność i wydajność w transporcie glukozy. Oprócz N-glikozylacji GLUT1 podlega również O-glikozylacji. Obie modyfikacje odgrywają główną rolę w osiąganiu pełnej aktywności transportowej i wpływają na masę molekularną białka [7,66]. Sugeruje się istnienie korelacji pomiędzy stopniem glikozylacji GLUT1, a złośliwością komórek nowotworowych. Zwiększoną masę molekularną GLUT1 osiągającą 70 kDa zaobserwowano w linii komórek nowotworowych CGL4, stanowiących hybrydę pochodzącą od komórek HeLa i fibroblastów człowieka, w porównaniu z linią niezesłośliwioną CGL1 wywodzącą się z tych samych komórek [56].
Wysoki stopień homologii pomiędzy poszczególnymi transporterami GLUT jest zdeterminowany obecnością licznych konserwatywnych ewolucyjnie sekwencji aminokwasowych. Maiden i wsp. [42] opisali pentameryczne sekwencje (RXGRR) umiejscowione w cytoplazmatycznych pętlach łączących segmenty 2 z 3 i 8 z 9 (ryc. 1). Ułożenie tych motywów w symetrycznych pozycjach obu połówek białek GLUT sugeruje, że transportery ssaków mogły powstać poprzez duplikacje odziedziczonego po przodkach 6 heliksowego transportera, chociaż niezależna ekspresja zarówno aminowej jak i karboksylowej części GLUT1 w komórkach Sf9 nie dawała funkcjonalnego białka [15]. Jednakże dodatni ładunek sekwencji RXGRR jest istotny dla prawidłowego wbudowania białek GLUT w błonę komórkową [68]. Podobnie motyw sekwencyjny zbudowany z czterech aminokwasów w regionie C-końcowym GLUT1 (DSQV) odpowiada za wiązanie domeny PDZ prezentowanej przez białka określane jako GIPC (Gα interacting protein, C-terminus). Stymulowane czynnikami wzrostu oddziaływanie GLUT1 z białkami GIPC promuje translokację transportera do błony komórkowej oraz zapobiega jego wewnątrzkomórkowej degradacji w lizosomach [94]. Inny konserwatywny motyw QLS zlokalizowany w domenie 7 transporterów GLUT1, 3 i 4 przypuszczalnie pełni rolę molekularnego filtra, który wpływa na swoistość substratową tych białek. Białka GLUT2 i 5, które transportują fruktozę mają odpowiednie motywy HVA i MGG. Eksperymenty mutacyjne oparte na przemiennej substytucji sekwencji QLS i HVA w transporterach GLUT3 i GLUT2 skutkowały częściowym odwróceniem ich kinetyki i selektywności substratowej [71]. Mutacje w genie SLC2A1 o charakterze zmiany ramki odczytu, zmiany sensu w obrębie aminokwasów zaangażowanych w tworzenie miejsca wiązania substratu lub całkowity brak ekspresji białka na skutek utraty heterozygotyczności prowadzi do metabolicznego zespołu niedoboru białkowego transportera 1 (choroba De Vivo) (GLUT1-deficiency syndrome – GLUT1-DS) dziedziczonego w sposób autosomalny dominujący. U wszystkich pacjentów z zespołem De Vivo występuje niskie stężenie glukozy w płynie mózgowo-rdzeniowym, którego następstwem są objawy epileptyczne, opóźniony rozwój psychoruchowy oraz ataksja [6].
Mechanizm transportu z udziałem GLUT1
Strukturalny model transportera GLUT1 zakłada formowanie centralnie położonego kanału wodnego utworzonego przez zestawienie pięciu transmembranowych α-helis [3,5,7,10,11], z których każda ma charakter amfipatyczny (ryc. 1). Istotną rolę w tworzeniu wodnego rdzenia odgrywają niepolarne reszty aminokwasowe oddziałujące wiązaniami hydrofobowymi z cząsteczkami wody [26,39]. Interakcje aminokwasów z płaszczem wodnym umożliwiają transporterowi GLUT1 funkcjonowanie w metastabilnych warunkach, w których mała zmiana energii wywołana przyłączeniem substratu, inhibitorów lub ATP powoduje duże zmiany w konformacji białka. Dlatego mutacje hydrofobowych reszt aminokwasowych GLUT1 w obrębie kanału prawdopodobnie oddziaływają z cząsteczkami wody prowadząc do utraty pełnej funkcjonalności transportera [32,40,44].
Początkowo w opisie mechanizmu transportu z udziałem GLUT1 sugerowano istnienie pojedynczego zewnętrznego i wewnętrznego miejsca wiązania substratu, które nie mogą absorbować liganda w tym samym czasie [3]. Wskazywano również na jednoczesne współdziałanie kilku miejsc wiązania liganda po obu stronach błony plazmatycznej. Niedawno przeprowadzone badania sugerują, że GLUT1 ma przynajmniej dwa zewnątrz- i wewnątrzkomórkowe miejsca wiązania glukozy połączone wspólnym kanałem wodnym [14,63]. Oparta na zasadach termodynamiki hipoteza transportu zaproponowana przez Naftalina [53] zakłada przyspieszoną wymianę cukru pomiędzy miejscami wiązania liganda leżącymi po przeciwnych stronach kanału w procesie tzw. „podwójnej wymiany” (germinate exchange), w którym zarówno wewnętrzne jak i zewnętrzne miejsca wiązania liganda absorbują cukier w tym samym czasie.
Porównanie transportu kilku analogów heksoz jako substratów oraz inhibitorów kompetycyjnych GLUT1 pozwoliło na wyciągnięcie wniosku, że wiązanie D-glukozy wymaga utworzenia kilku wiązań wodorowych pomiędzy grupami hydroksylowymi w pozycjach C1, C2 i C3 cukru, a polarnymi resztami zewnętrznego miejsca wiązania liganda [10,27]. Prawdopodobnie w przyłączeniu D-glukozy do GLUT1 uczestniczy również grupa hydroksylowa przy C4, ponieważ epimer D-glukozy, D-galaktoza ma 10-krotnie mniejsze powinowactwo do GLUT1 niż D-glukoza [62].
Badania wskazują na występowanie białek GLUT1 w postaci dimerów. Dimery te tworzą tetramer, w których obie podjednostki działają kooperatywnie tworząc kompleks nieustanie prezentujący przynajmniej dwa zewnętrzne i dwa wewnętrzne miejsca wiązania substratu [100].
Ze względu na brak krystalograficznej analizy struktury białka GLUT1 trudno jest na podstawie istniejących modeli i doniesień literaturowych jednoznacznie odpowiedzieć na pytanie w jaki sposób transporter glukozy 1 przeprowadza translokację substratu przez błonę komórkową. Jednak mimo braku pełnej informacji o trzeciorzędowej strukturze GLUT1, obecna wiedza może umożliwić zrozumienie, a w konsekwencji leczenie pacjentów z chorobami związanymi z homeostazą glukozy.
Nadekspresja GLUT1 w nowotworach
Wysoki poziom ekspresji GLUT1 zaobserwowano w wielu typach nowotworów. W rakach piersi i jelita grubego ekspresja GLUT1 ulegała podwyższeniu wraz ze wzrostem stopnia zaawansowania nowotworu i gorszym rokowaniem dla pacjenta [4,12,61]. W nowotworach gruczołów ślinowych ujawniono związek pomiędzy poziomem ekspresji GLUT1, a rozmiarem guza i zdolnością komórek do metastazy [51]. W rakach żołądka ekspresja GLUT1 ściśle korelowała z typem nowotworu, angiogenezą oraz przerzutowaniem do węzłów chłonnych i wątroby [33]. Wyciszenie ekspresji genu GLUT1 w komórkach białaczkowych człowieka HL-60 prowadziło do zahamowania proliferacji komórek o ponad 50% [10]. Badania przeprowadzone przez Noguchiego i wsp. [55] na linii komórkowej raka żołądka (MKN45) wykazały 85% spadek pobierania glukozy oraz zmiany w cyklu komórkowym ograniczające wzrost nowotworu na skutek zahamowania ekspresji GLUT1 po transfekcji wektorem kodującym antysensowne GLUT1 cDNA.
Wyniki dotychczasowych badań zgodnie sugerują zastosowanie GLUT1 jako prognostycznego wskaźnika złośliwości oraz stopnia zaawansowania nowotworu, który umożliwiłby wyodrębnienie pacjentów wymagających agresywniejszej terapii. Nadekspresja GLUT1 wynikająca ze zwiększonego zapotrzebowania komórek nowotworowych na energię promuje poprzez szlak glikolizy wzrost rozmiarów guza i jego inwazyjność [20]. Fenomen zwiększonego pobierania glukozy przez komórki nowotworowe ma kliniczne zastosowanie w diagnostyce wielu typów nowotworów złośliwych i ich przerzutów przez wprowadzenie znacznika [18F]-fluoro-2-deoksy-D-glukozy (FDG) w jednej z najbardziej efektywnych metod medycyny molekularnej, tj. pozytonowej tomografii emisyjnej (PET). Podwyższony poziom ekspresji GLUT1 oraz wysoka maksymalna standardowa wartość wychwytu 18FDG zaobserwowane w płaskonabłonkowych rakach płuc w porównaniu z gruczolakami, korelowały ze stopniem zróżnicowania komórek nowotworu oraz przerzutami do węzłów chłonnych [88]. Podobnie w guzach grasicy pochodzenia nabłonkowego wychwyt 18FDG był ściśle związany z poziomem ekspresji GLUT1 i heksokinazy II [54]. Aktywny transport z udziałem GLUT1 wymaga jego translokacji do błony komorkowej. W liniach komórkowych mięsaków kościopochodnych zaobserwowano zwiększone pobieranie glukozy wynikające z przemieszczenia GLUT1 do błony komórkowej w odpowiedzi na stymulację komórek insuliną [13]. Inne badania przeprowadzone na linii komórkowej białaczki megakariocytowej człowieka sugerują translokację GLUT1 z przedziałów wewnątrzkomórkowych do błony plazmatycznej pod wpływem czynnika wzrostowego komórek pnia (stem cell factor – SCF) oraz H2O2 [45]. Regulacja ekspresji GLUT1 w komórkach nowotworowych jest procesem wieloczynnikowym, w który zaangażowane są m.in.: hipoksja [24], hormony [19], czynniki wzrostu [16], geny supresorowe [70], onkogeny [97].
Rola szlaku PI3K/AKT/mTOR w ekspresji GLUT1
Aktywacja kinazy mTOR odgrywa rolę w przebiegu wielu przemian promujących transformację nowotworową. Białko mTOR zaangażowane w liczne szlaki sygnalizacyjne pośredniczy w regulacji metabolizmu, wzrostu komórki, proliferacji i angiogenezy [99]. Badania wykazały, że w wielu nowotworach człowieka PI3K/AKT – zależna regulacja kinazy mTOR koreluje z poziomem ekspresji transportera GLUT1 [77]. Aktywacja kaskady PI3K/AKT/mTOR zachodzi w wyniku związania insuliny lub czynników wzrostu przez receptory o charakterze kinaz tyrozynowych. Przyłączenie liganda powoduje ich autofosforylację i rekrutację do błony komórkowej 3-kinazy fosfatydyloinozytolu (PI3K), która przekształca fosfatydyloinozytolo-4,5-bisfosforan (PIP2) do fosfatydyloinozytolo-3,4,5-trifosforanu (PIP3) (ryc. 2). PIP3 wiąże się w obrębie domeny PH serynowo-treoninowej kinazy AKT promując jej translokację do błony. Aktywacja AKT1 wymaga fosforylacji Thr 308 i Ser 473 umiejscowionych odpowiednio w domenie kinazowej i regulatorowej. Za fosforylację Thr 308 odpowiada kinaza PDK1 rekrutowana do błony komórkowej przez PIP3. W fosforylację Ser 408 mogą być zaangażowane różne kinazy: PDK1, PDK2, ILK, DNA-PK, mTORC2 lub proces ten przebiega autokatalitycznie [35]. AKT fosforyluje wiele substratów, spośród których dwa TSC2 (tuberous sclerosis 2) i PRAS40 (proline-rich AKT-substrate-40) prowadzą do bezpośredniej aktywacji serynowo-treoninowej kinazy mTOR [80]. W komórce mTOR wchodzi w skład dwóch niezależnych kompleksów odmiennych pod względem funkcji, regulacji i wrażliwości na rapamycynę. W skład mTORC1 pośredniczącego w regulacji procesów wzrostu i odżywiania komórkowego wchodzą białka: mTOR, mLST8, Raptor, podczas gdy główne składowe mTORC2 stanowią: mTOR, mLST8, Rictor, mSin1, Protor i PRR5. AKT-zależny szlak sygnalizacji przebiega z udziałem kompleksu mTORC1. Białko TSC2 o aktywności GTP-azowej w połączeniu z TSC1 reguluje aktywność GTP-azy Rheb. Fosforylacja Ser 939 i Thr 1462 w TSC2 hamuje GTP-azową aktywność heterodimeru TSC1/TSC2, co prowadzi do akumulacji białka Rheb połączonego z GTP, które w wyniku niewyjaśnionego mechanizmu aktywuje kinazę mTOR w kompleksie mTORC1 [43,64]. Początkowo uważano, że aktywacja mTOR jest wynikiem asocjacji Rheb-GTP z białkiem FKBP38 o funkcji supresorowej w stosunku do mTORC1 [86]. Obecnie przypuszcza się, że GTP-aza Rheb może stymulować fosfolipazę D do tworzenia kwasu fosfatydowego bezpośrednio modulującego aktywność kinazy mTOR [83]. Mechanizm aktywacji mTORC1 przez GTP-azę Rheb wymaga farnezylacji białka Rheb i może być blokowany przez inhibitory transferazy farnezylowej [9].

Ryc. 2. Szlaki sygnalizacyjne mTOR regulujące ekspresję GLUT1 (opis w tekście)
Drugi sposób aktywacji mTORC1 przez AKT polega na fosforylacji białka PRAS40 – negatywnego regulatora kinazy mTOR, działającego niezależnie od TSC2. Fosforylacja w PRAS40 Thr 246 prowadzi do jego oddysocjowania od kompleksu mTORC1 i aktywacji kinazy mTOR. Jednocześnie PRAS40 sam stanowi substrat dla mTORC1, kontrolującego własną aktywność sygnalizacyjną poprzez fosforylację reszt seryny swojego supresora [92]. Zależności pomiędzy kinazami AKT i mTOR dodatkowo komplikuje kompleks mTORC2, który w pewnych komórkach nie wykazuje wrażliwości na rapamycynę. Kompleks mTORC2 potrafi bezpośrednio fosforylować Ser 473 AKT w odpowiedzi na stymulację czynnikami wzrostu [27]. Utworzenie kompleksu mTORC1 hamuje formowanie kompleksu mTORC2 i ogranicza możliwość aktywacji AKT. Sygnalizacja z udziałem AKT i mTOR jest ze sobą powiązana wzajemnym sprzężeniem zwrotnym wykluczającym możliwość ciągłej hiperaktywacji obu kinaz [79].
Do najlepiej poznanych efektorów aktywności mTOR należą kinaza białkowa p70S6 oraz białko 4E-BP1 zaangażowane w proces inicjacji translacji. Większość dojrzałych eukariotycznych mRNA ma na końcu 5′ czapeczkę zbudowaną z 7-metyloguanozyny (m7G), a na końcu 3′ sekwencję poli(A). Inicjacja translacji wymaga rozpoznania czapeczki przez czynnik eIF4E, którego aktywność jest hamowana przez związanie ze swoistym inhibitorem białkowym 4E-BP1. W wyniku fosforylacji 4E-BP1 przez mTOR dochodzi do aktywacji eIF4E i tworzenia kompleksu inicjacyjnego [11,22]. Badania kinazy p70S6 sugerują jej udział w fosforylacji podjednostki rybosomu 40S i związanej z tym zwiększonej wydajności translacji populacji mRNA mających na swoim końcu 5′ łańcuch polipirymidynowy (5′ TOP mRNA). Są to głównie transkrypty kodujące białka rybosomalne oraz czynniki translacyjne np. eEF1A. Fosforylacja p70S6 przez mTOR aktywuje białko S6, które zwiększa efektywność translacji 5’TOP mRNA [28]. Ostatnie doniesienia wskazują, że sygnalizacja mTOR-p70S6 może również kontrolować inne czynniki translacyjne – eIF3 oraz eIF4B [75].
Obecnie sugeruje się istnienie związku pomiędzy nadekspresją GLUT1 i aktywacją szlaku sygnalizacyjnego z udziałem kompleksu mTORC1. Jednak do tej pory nie udało się wyjaśnić w jakim stopniu kinaza mTOR bezpośrednio wpływa na regulację translacji białka GLUT1. Możliwe, że zwiększenie wydajności translacji innych białek pośrednio wpływa na regulację ekspresji GLUT1 w komórkach nowotworowych. Co więcej, kinaza mTOR może wchodzić w interakcje z różnymi białkami np.: Rac1, TELO2 i FXBW7 inhibitorami kinaz zależnymi od cyklin p21 i p27, kinazą białkową C [93]. Obecnie niewiele wiadomo o ekspresji genów i białek kontrolowanych przez kinazę mTOR, choć ich liczba systematycznie rośnie.
Regulacja ekspresji GLUT1 przez czynnik transkrypcyjny HIF-1
Niedotlenienie nowotworu powoduje aktywację czynnika transkrypcyjnego HIF-1, pośredniczącego w regulacji ekspresji wielu genów, których produkty białkowe uczestniczą w szlakach przekazywania sygnału związanych z metabolizmem glukozy, angiogenezą i inwazją komórek nowotworowych. Białko HIF-1 funkcjonuje jako heterodimer składający się z podjednostek HIF-1α i HIF-1β [2] (ryc. 3). W warunkach fizjologicznych podjednostka HIF-1α podlega hydroksylacji przez hydroksylazę prolinową (HPH), której aktywność jest uwarunkowana obecnością tlenu w tkankach. Hydroksylacja reszt Pro 402 i 564 umożliwia wiązanie HIF-1 z białkiem vHL rozpoznawanym przez ligazę ubikwitynową, co prowadzi do degradacji powstałego kompleksu w proteasomie [59]. Oprócz reszt proliny, hydroksylacja Asn 803 przez hydroksylazę FIH-1 osłabia interakcje HIF-1α z białkami p300 i CBP – kofaktorami zaangażowanymi w regulację ekspresji genów. Zarówno HPH jak i FIH-1 do pełnej aktywności enzymatycznej wymagają cząsteczki tlenu i żelaza. Ponadto w regulacji aktywności i stabilności HIF-1 uczestniczy acetylacja i fosforylacja [29].
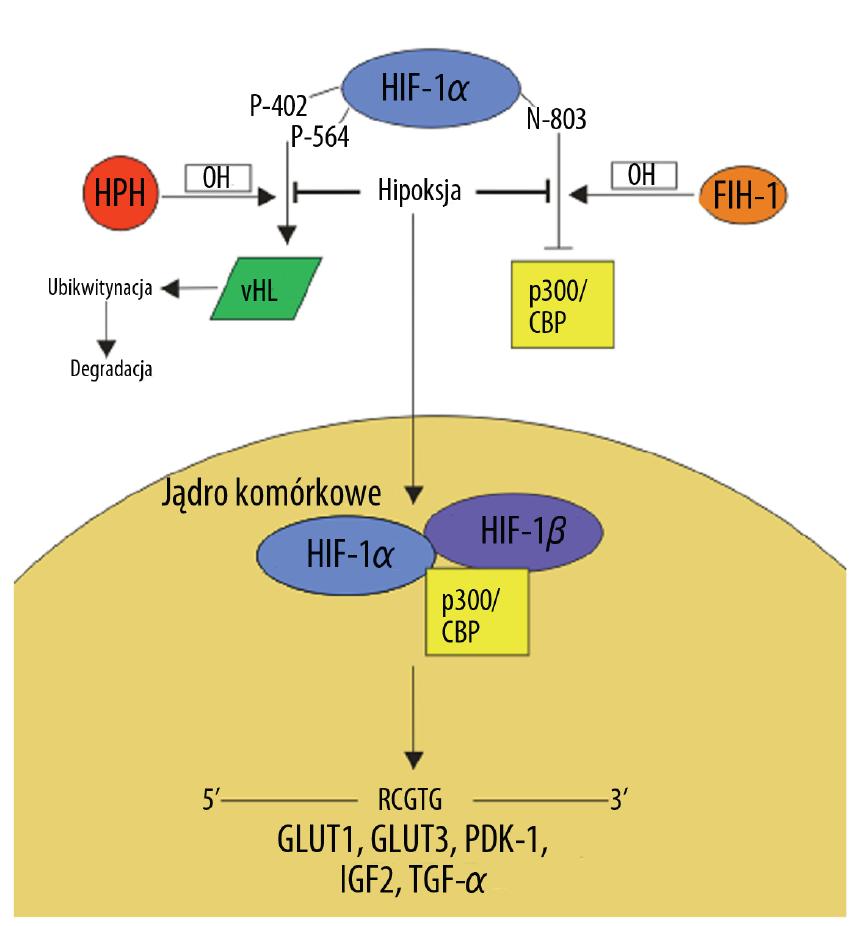
Ryc. 3. Regulacja ekspresji GLUT1 przez HIF-1 (opis w tekście)
Hipoksja jest podstawową przyczyną nadekspresji HIF-1 w komórkach nowotworowych. Niedobór tlenu inaktywuje enzymy odpowiedzialne za hydroksylację HIF-1, która jest konieczna do tworzenia kompleksu z vHL i ubikwitynozależnej degradacji HIF-1α w proteasomach. W warunkach tlenowych ograniczony stopień degradacji HIF-1α wynika głównie z mutacji, utraty lub obniżonej ekspresji supresora nowotworowego vHL. Pozostałe mechanizmy nadekspresji HIF-1 dotyczą regulacji syntezy podjednostki α [30]. W wielu typach nowotworów stymulacja transkrypcji i translacji HIF-1α wynika ze zwiększonej ekspresji receptorów o aktywności kinaz tyrozynowych uruchamiających kaskadę przekazywania sygnału z udziałem szlaku PI3K/AKT [74]. Podobnie nasiloną ekspresję HIF-1α zaobserwowano w guzach z mutacją w białku TSC – supresorze kinazy mTOR promującej progresję nowotworową poprzez zintensyfikowanie wydajności translacji wielu białek [25]. Ponadto HIF-1 może sam pośrednio wpływać na poziom ekspresji HIF-1α poprzez stymulację transkrypcji czynników wzrostu, takich jak: IGF2 (insulin-like growth factor) i TGF-α (transforming growth factor), które uruchamiają szlaki przekazywania sygnału związane z progresja guza [38].
W wyniku przedstawionych wyżej mechanizmów dochodzi do akumulacji podjednostki HIF-1α w cytoplazmie komórek nowotworowych. Następnie HIF-1α migruje do jądra komórkowego, gdzie łączy się z konstytutywnie stabilną i tlenowo niezależną podjednostką HIF-1β. Powstały heterodimer stanowi aktywną postać czynnika transkrypcyjnego HIF-1, który wraz z swoimi kofaktorami p300 i CBP rozpoznaje sekwencję 5′-RCGTG-3′ tzw. HRE (hypoxia – response elements) zlokalizowaną w obrębie promotorów i wzmacniaczy wielu genów regulujących procesy proliferacji, apoptozy, angiogenezy i metabolizmu komórkowego [50].
Badania przeprowadzone na tkankach oraz liniach komórkowych wskazują na ścisłą zależność poziomu ekspresji transporterów glukozy GLUT1 i GLUT3 od stopnia aktywacji HIF-1. Czynnik ten indukuje transkrypcję genów GLUT1 i GLUT3 oraz enzymów uczestniczących w procesie glikolizy, nasilając tym samym tempo metabolizowania glukozy [24,47]. Aktywacja HIF-1 w komórkach nowotworowych prowadzi do trwałego wygaszenia oddychania mitochondrialnego i przesunięcia metabolizmu glukozy w stronę glikolizy. Zmiany te wynikają z bezpośredniej stymulacji ekspresji mitochondrialnej kinazy dehydrogenazy pirogronianowej, która inaktywuje dehydrogenazę pirogronianową (PDH). Inaktywacja PDH ogranicza przekształcanie pirogronianu do acetylo-CoA – głównego metabolitu cyklu Krebsa [72]. Ponadto warunki hipoksji nowotworu prowadzą do negatywnej regulacji heterogennych jądrowych rybonukleoprotein A2 i L (hnRNP A2 i hnRNP L), które wiążąc się z sekwencją AURE (AU-rich response element) w obrębie regionu 3’UTR (untranslated region) zwiększają niestabilność GLUT1 mRNA i blokują jego translację [23]. Nasilone pobieranie glukozy przez komórki nowotworowe stanowi alternatywę pozwalającą zaspokoić głód energetyczny wynikający z niewydajnego oddychania beztlenowego.
Aktywacja ekspresji GLUT1 przez onkogeny
Onkogeny powstają w wyniku mutacji w obrębie protoonkogenów. Ekspresja onkogenów reguluje poziom syntezy lub zmienia właściwości białek zaangażowanych w regulację wzrostu, różnicowania i podziału komórki. Jednymi z najlepiej poznanych białek pośredniczących w przekazywaniu sygnału ze środowiska zewnątrzkomórkowego do jądra komórkowego są białka Ras uczestniczące w aktywacji kaskady Raf/MEK/ERK [69]. Mutacje w onkogenie RAS występują w 95% przypadków raków trzustki, 50% przypadków raków jelita grubego oraz niektórych typach nowotworów płuc [57]. Mutacje w KRAS wykluczają mutacje w onkogenie BRAF rozpowszechnione w czerniakach oraz rakach jelita grubego bez mutacji w KRAS [17]. Nadekspresja lub zwiększona aktywność białek Ras powoduje nieustanną stymulację szlaku sygnalizacyjnego kinaz aktywowanych mitogenem (MAPK) [36]. Aktywacja kinaz ERK może prowadzić do zwiększonej aktywności kinazy mTOR w wyniku hamowania właściwości supresorowych białka TSC2 [65] (ryc. 2). Badania przeprowadzone na liniach komórkowych raka jelita grubego o zróżnicowanym statusie mutacyjnym w onkogenach KRAS i BRAF wykazały ścisłą korelację tych mutacji z poziomem transkrypcji transportera GLUT1. Zaobserwowano wzrost ekspresji GLUT1 (3-22-krotny) w komórkach z mutacjami w allelach KRAS i BRAF w porównaniu z allelami o fenotypie dzikim. Ponadto nie stwierdzono związku pomiędzy nadekspresją transkryptu GLUT1, a poziomem aktywności HIF-1α [97]. Jednak, wyniki badań przeprowadzonych na nowotworach okrężnicy ujawniły znacznie niższą (30%) częstość mutacji punktowych w kodonie 12 KRAS w porównaniu z ekspresją GLUT1 (100%) [55]. Inne doniesienia sugerują, że nadekspresja GLUT1 w fibroblastach wymaga aktywacji onkogenu c-Fos. Białka Fos i Jun wchodzą w skład heterodimeru AP-1, który wiąże się z sekwencją TRE rozpoznawaną przez estry forbolu, regulując ekspresję wielu genów. Obecnie mimo braku przekonujących dowodów przypuszcza się, że w obrębie regionów wzmacniaczy genu SLC2A1 kodującego GLUT1 mogą występować motywy TRE regulowane przez AP-1 [67]. Podobne sekwencje wzmacniaczy aktywowane przez onkogen v-src zidentyfikowano w regionie promotorowym genu SLC2A1 u myszy [52]. Badania przeprowadzone na fibroblastach szczura Rat1, kodujących białko fuzyjne łączące Myc z domeną wiązania liganda receptora estrogenowego (MycER) wykazały, że geny kodujące GLUT1, fosfofruktokinazy i enolazy mogą być bezpośrednio regulowane przez onkogen c-myc [58]. W innym modelu badawczym onkogen c-myc przez wiązanie z sekwencją zlokalizowaną w obrębie miejsca promotorowego prowadził do bezpośredniej nadekspresji genu kodującego dehydrogenazę mleczanową (LDH-A) [76].
GLUT1 w terapii przeciwnowotworowej
Zwiększone pobieranie glukozy i związane z nim przyspieszone tempo glikolizy wynika z nadekspresji oraz translokacji transporterów glukozy do błony komórkowej. Z tego względu białka GLUT, a zwłaszcza GLUT1, którego nadekspresję w nowotworach obserwuje się najczęściej, stały się potencjalnym celem terapii przeciwnowotworowej. Regulacja GLUT1 może stanowić główny czynnik wpływający na metabolizm glukozy w odpowiedzi na chemioterapię.
Większość zidentyfikowanych do tej pory inhibitorów aktywności GLUT1 działa głównie na zasadzie współzawodnictwa o miejsce wiązania substratu. Na przykład cytochalasyna B – przenikający przez błonę komórkową alkaloid – niszczy filamenty aktynowe i hamuje transport glukozy [5]. Kolejny związek o budowie diterpenu – forskolina po przedostaniu się do cytoplazmy blokuje transporter GLUT1 i aktywuje cyklazę adenylanową [73]. Podobnymi właściwościami charakteryzują się: izoflawonoid genisteina, gossypol, florentyna chalkonowa [5,60,90]. Obecnie rośnie zainteresowanie właściwościami D-allozy, rzadko spotykanego w naturze analogu D-glukozy. Mitani i wsp. [49] w badaniach przeprowadzonych na trzech liniach komórkowych nowotworów głowy i szyi wykazali, że cukier ten jest kompetycyjnym inhibitorem transportu glukozy do wnętrza komórki. Komórki traktowane D-allozą charakteryzowały się obniżonym poziomem ekspresji cyklin A2, B1 i kinazy CDC2 oraz podwyższonym poziomem syntezy p21 i p53 mRNA. Podobne wyniki uzyskali Sui i wsp. [82], którzy hodując komórki nowotworowe wątroby na podłożu zawierającym D-allozę zaobserwowali nadekspresję p21 i p27 oraz związane z tym zahamowanie cyklu komórkowego w fazie G2/M i uruchomienie apoptozy.
Znane są także pośrednie inhibitory GLUT1 skierowane przeciwko szlakom sygnalizacyjnym regulującym poziom ekspresji GLUT1. Zaobserwowano m.in. spadek syntezy GLUT1 w komórkach raka trzustki traktowanych apigeniną – flawonoidem roślinnym o działaniu supresorowym w stosunku do szlaku PI3K/Akt [48].
Jednak nadekspresja transporterów GLUT umożliwia akumulację w komórkach nowotworowych pochodnych glukozy o charakterze terapeutycznym. W wielu badaniach nad wykorzystaniem 2-deoksyglukozy (2-DG) w terapii wykazano, że związek ten jest kompetycyjnym inhibitorem procesu glikolizy [41]. W wyniku transportu do wnętrza komórki 2-DG ulega fosforylacji przez heksokinazę, ale w przeciwieństwie do glukozo-6-fosforanu nie jest substratem dla izomerazy glukozofosforanowej. Nagromadzenie 2-DG-P w komórce prowadzi do zahamowania procesu glikolizy, a następnie zatrzymania cyklu komórkowego i uruchomienia apoptozy [1]. Ponadto ostatnie doniesienia sugerują, że w przypadku guzów rosnących w warunkach normoksji 2-DG jest skutecznym inhibitorem N-glikozylacji, niezbędnej do translokacji GLUT1 do błony komórkowej [37]. W badaniach in vivo przeprowadzonych na myszach z przeszczepionym mięsakiem kości lub niedrobnokomórkowym rakiem płuc człowieka 2-DG uwrażliwiała komórki nowotworowe na chemioterapię adriamycyną i paklitakselem [46]. Co więcej, eksperymenty kliniczne przeprowadzone z udziałem pacjentów z glejakiem wielopostaciowym wykazały, że stosowanie 2-DG w dawkach bezpiecznych, tj. do 250 mg/kg masy ciała uczula komórki nowotworu na radioterapię [78]. Podobnie podawanie 2-DG w zestawieniu z 6-aminonikotynamidem (inhibitor dehydrogenazy glukozo-6-fosforanu) obniżało komórkowe stężenie glutationu i zwiększało śmiertelność linii komórek glejaka pod wpływem czynnika radiacyjnego [89]. Oprócz łączonej terapii z 2-DG, rośnie zainteresowanie syntezą koniugatów konwencjonalnych cytotoksycznych leków z resztami glukozy, które mogłyby zwiększyć dostępność chemioterapii do wnętrza guza. Przykładem takiego związku jest 2-GluSNAP, połączenie glukozy z donorem tlenku azotu S-nitroso-N-acetyl-penicillaminą (SNAP). Stwierdzono, że nasilenie cytotoksycznego działania 2-GluSNAP w komórce koreluje wraz ze wzrostem poziomu ekspresji transportera GLUT1 [81]. Zastosowanie kliniczne niemetabolizowalnych analogów glukozy wydaje się korzystne dlatego, iż w przeciwieństwie do komórek prawidłowych, które potrafią metabolizować różne cukry, głównym substratem energetycznym dla komórek nowotworowych jest glukoza. Obecnie prowadzone są badania kliniczne nad bezpieczeństwem i skutecznością działania tych związków jako przyszłych terapeutyków [8].
Podsumowanie
Komórki nowotworowe przearanżowują swój metabolizm w kierunku procesu beztlenowej glikolizy. Zwiększone tempo glikolizy wymaga nasilenia transportu glukozy do wnętrza komórki, które w znacznym stopniu wynika z nadekspresji transportera GLUT1. Przeprowadzone do tej pory badania jednoznacznie podkreślają znaczenie GLUT1 w progresji i uzłośliwieniu komórek nowotworowych.
W wielu typach nowotworów ekspresja GLUT1 koreluje ze stadium zaawansowania choroby i gorszym rokowaniem dla pacjenta stając się potencjalnym czynnikiem prognostycznym. Jednak nadekspresja GLUT1 może zwiększać czułość detekcji nowotworów techniką 18FDG-PET oraz uczulać pacjentów na stosowaną powszechnie chemio- i radioterapię. W przyszłości dokładniejsze poznanie struktury i regulacji GLUT1 w komórce przyczyni się do lepszego zrozumienia i leczenia chorób związanych z homeostazą glukozy.
PIŚMIENNICTWO
[1] Aft R.L., Zhang F.W., Gius D.: Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. Br. J. Cancer, 2002; 87: 805-812
[PubMed] [Full Text HTML] [Full Text PDF]
[2] Airley R.E., Mobasheri A.: Hypoxic regulation of glucose transport, anaerobic metabolism and angiogenesis in cancer: novel pathways and targets for anticancer therapeutics. Chemotherapy, 2007; 53: 233-256
[PubMed]
[3] Appleman J.R., Lienhard G.E.: Kinetics of the purified glucose transporter. Direct measurement of the rates of interconversion of transporter conformers. Biochemistry, 1989; 28: 8221-8227
[PubMed]
[4] Ayala F.R., Rocha R.M., Carvalho K.C., Carvalho A.L., da Cunha I.W., Lourenço S.V., Soares F.A.: Glut1 and Glut3 as potential prognostic markers for oral squamous cell carcinoma. Molecules, 2010; 15: 2374-2387
[PubMed]
[5] Bloch R.: Inhibition of glucose transport In the human erythrocyte by cytochalasin B. Biochemistry, 1973; 12: 4799-4801
[PubMed]
[6] Brockmann K.: The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev., 2009; 31: 545-552
[PubMed]
[7] Buse M.G., Robinson K.A., Marshall B.A., Hresko R.C., Mueckler M.M.: Enhanced O-GlcNAc protein modification is associated with insulin resistance in GLUT1-overexpressing muscles. Am. J. Physiol. Endocrinol. Metab., 2002; 283: E241-E250
[PubMed] [Full Text HTML] [Full Text PDF]
[8] Calvo M.B., Figueroa A., Pulido E.G., Campelo R.G., Aparicio L.A.: Potential role of sugar transporters in cancer and their relationship with anticancer therapy. Int. J. Endocrinol., 2010; 2010: 205357
[PubMed] [Full Text HTML] [Full Text PDF]
[9] Castro A.F., Rebhun J.F., Clark G.J., Quilliam L.A.: Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J. Biol. Chem., 2003; 278: 32493-32496
[PubMed] [Full Text HTML] [Full Text PDF]
[10] Chan J.Y., Kong S.K., Choy Y.M., Lee C.Y., Fung K.P.: Inhibition of glucose transporter gene expression by antisense nucleic acids in HL-60 leukemia cells. Life Sci., 1999; 65: 63-70
[PubMed]
[11] Choo A.Y., Yoon S.O., Kim S.G., Roux P.P., Blenis J.: Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA, 2008; 105: 17414-17419
[PubMed] [Full Text HTML] [Full Text PDF]
[12] Chung F.Y., Huang M.Y., Yeh C.S., Chang H.J., Cheng T.L., Yen L.C., Wang J.Y., Lin S.R.: GLUT1 gene is a potential hypoxic marker in colorectal cancer patients. BMC Cancer, 2009; 9: 241
[PubMed] [Full Text HTML] [Full Text PDF]
[13] Cifuentes M., García M.A., Arrabal P.M., Martínez F., Yanez M.J., Jara N., Weil B., Domínguez D., Medina R.A., Nualart F.: Insulin regulates GLUT1-mediated glucose transport In MG-63 human osteosarcoma cells. J. Cell. Physiol., 2011; 226: 1425-1432
[PubMed]
[14] Cloherty E.K., Levine K.B., Carruthers A.: The red blood cell glucose transporter presents multiple, nucleotide-sensitive sugar exit sites., Biochemistry, 2001; 40: 15549-15561
[PubMed]
[15] Cope D.L., Holman G.D., Baldwin S.A., Wolstenholme A.J.: Domain assembly of the GLUT1 glucose transporter. Biochem J., 1994; 300: 291-294
[PubMed] [Full Text HTML] [Full Text PDF]
[16] Díaz M., Vraskou Y., Gutiérrez J., Planas J.V.: Expression of rainbow trout glucose transporters GLUT1 and GLUT4 during in vitro muscle cell differentiation and regulation by insulin and IGF-I. Am. J. Physiol. Regul. Integr. Comp. Physiol., 2009; 296: R794-R800
[PubMed] [Full Text HTML] [Full Text PDF]
[17] Downward J.: Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer, 2003; 3: 11-22
[PubMed]
[18] Ferreira L.M.R.: Cancer metabolism: the Warburg effect today. Exp. Mol. Pathol., 2010; 89: 372-380
[PubMed]
[19] Frolova A., Flessner L., Chi M., Kim S.T., Foyouzi-Yousefi N., Moley K.H.: Facilitative glucose transporter type 1 is differentially regulated by progesterone and estrogen in murine and human endometrial stromal cells. Endocrinology, 2009; 150: 1512-1520
[PubMed] [Full Text HTML] [Full Text PDF]
[20] Furuta E., Okuda H., Kobayashi A., Watabe K.: Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim. Biophys. Acta, 2010; 1805: 141-152
[PubMed] [Full Text HTML] [Full Text PDF]
[21] Ganapathy V., Thangaraju M., Prasad P.D.: Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol. Ther., 2009; 121: 29-40
[PubMed]
[22] Grolleau A., Bowman J., Pradet-Balade B., Puravs E., Hanash S., Garcia-Sanz J.A., Beretta L.: Global and specific translational control by rapamycin in T cells uncovered by microarrays and proteomics. J. Biol. Chem., 2002; 277: 22175-22184
[PubMed] [Full Text HTML] [Full Text PDF]
[23] Hamilton B.J., Nichols R.C., Tsukamoto H., Boado R.J., Pardridge W.M., Rigby W.F.: hnRNP A2 and hnRNP L bind the 3’UTR of glucose transporter 1 mRNA and exist as a complex in vivo. Biochem. Biophys. Res. Commun., 1999; 261: 646-651
[PubMed]
[24] Hayashi M., Sakata M., Takeda T., Yamamoto T., Okamoto Y., Sawada K., Kimura A., Minekawa R., Tahara M., Tasaka K., Murata Y.: Induction of glucose transporter 1 expression through hypoxia-inducible factor 1α under hypoxic conditions in trophoblast-derived cells. J. Endocrinol., 2004; 183: 145-154
[PubMed] [Full Text HTML] [Full Text PDF]
[25] Hebert C., Norris K., Parashar P., Ord R.A., Nikitakis N.G., Sauk J.J.: Hypoxia-inducible factor-1α polymorphisms and TSC1/2 mutations are complementary in head and neck cancers. Mol. Cancer, 2006; 5: 3
[PubMed] [Full Text HTML] [Full Text PDF]
[26] Hruz P.W., Mueckler M.M.: Structural analysis of the GLUT1 facilitative glucose transporter (review). Mol. Membr. Biol., 2001; 18: 183-193
[PubMed]
[27] Ikenoue T., Inoki K., Yang Q., Zhou X., Guan K.L.: Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J., 2008; 27: 1919-1931
[PubMed] [Full Text HTML] [Full Text PDF]
[28] Jefferies H.B., Reinhard C., Kozma S.C., Thomas G.: Rapamycin selectively represses translation of the “polypyrimidine tract” mRNA family. Proc. Natl. Acad. Sci. USA, 1994; 91: 4441-4445
[PubMed] [Full Text HTML] [Full Text PDF]
[29] Jeong J.W., Bae M.K., Ahn M.Y., Kim S.H., Sohn T.K., Bae M.H., Yoo M.A., Song E.J., Lee K.J., Kim K.W.: Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell, 2002; 111: 709-720
[PubMed] [Full Text HTML] [Full Text PDF]
[30] Jokilehto T., Jaakkola P.M.: The role of HIF prolyl hydroxylases in tumour growth. J. Cell. Mol. Med., 2010; 14: 758-770
[PubMed]
[31] Joost H.G., Thorens B.: The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members. Mol. Membr. Biol., 2001; 18: 247-256
[PubMed]
[32] Kasahara T., Maeda M., Boles E., Kasahara M.: Identification of a key residue determining substrate affinity in the human glucose transporter GLUT1. Biochim. Biophys. Acta, 2009; 1788: 1051-1055
[PubMed]
[33] Kawamura T., Kusakabe T., Sugino T., Watanabe K., Fukuda T., Nashimoto A., Honma K., Suzuki T.: Expression of glucose transporter-1 in human gastric carcinoma: association with tumor aggressiveness, metastasis, and patient survival. Cancer, 2001; 92: 634-641
[PubMed]
[34] KC S., Cárcamo J.M., Golde D.W.: Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J., 2005; 19: 1657-1667
[PubMed] [Full Text HTML] [Full Text PDF]
[35] Krześlak A.: Kinaza Akt: kluczowy regulator metabolizmu i progresji nowotworów. Postępy Hig. Med. Dośw., 2010; 64: 490-503
[PubMed] [Full Text HTML] [Full Text PDF]
[36] Krzyżowska M., Świątek W., Fijałkowska B., Niemiałtowski M., Schollenberger A.: Rola kinaz MAP w odpowiedzi immunologicznej. Postępy Biol. Kom., 2009; 36: 295-308
[Abstract] [Full Text PDF]
[37] Kurtoglu M., Gao N., Shang J., Maher J.C., Lehrman M.A., Wangpaichitr M., Savaraj N., Lane A.N., Lampidis T.J.: Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol. Cancer Ther., 2007; 6: 3049-3058
[PubMed] [Full Text HTML] [Full Text PDF]
[38] Land S.C., Tee A.R.: Hypoxia-inducible factor 1 α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem., 2007; 282: 20534-20543
[PubMed] [Full Text HTML] [Full Text PDF]
[39] Levy Y., Onuchic J.N.: Water and proteins: a love-hate relationship. Proc. Natl. Acad. Sci. USA, 2004; 101: 3325-3326
[PubMed] [Full Text HTML] [Full Text PDF]
[40] Liu Q., Vera J.C., Peng H., Golde D.W.: The predicted ATP-binding domains in the hexose transporter GLUT1 critically affect transporter activity. Biochemistry, 2001; 40: 7874-7881
[PubMed]
[41] Maher J.C., Savaraj N., Priebe W., Liu H., Lampidis T.J.: Differential sensitivity to 2-deoxy-D-glucose between two pancreatic cell lines correlates with GLUT1 expression. Pancreas, 2005; 30: e34-e39
[PubMed]
[42] Maiden M.C., Davis E.O., Baldwin S.A., Moore D.C., Henderson P.J.: Mammalian and bacterial sugar transport protein are homologous. Nature, 1987; 325: 641-643
[PubMed]
[43] Manning B.D., Tee A.R., Logsdon M.N., Blenis J., Cantley L.C.: Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinisitide 3-kinase/Akt pathway. Mol. Cell, 2002; 10: 151-162
[PubMed] [Full Text HTML] [Full Text PDF]
[44] Manolescu A.R., Witkowska K., Kinnaird A., Cessford T., Cheeseman C.: Facilitated hexose transporters: new perspectives on form and function. Physiology. 2007; 22: 234-240
[PubMed] [Full Text HTML] [Full Text PDF]
[45] Maraldi T., Fiorentini D., Prata C., Landi L., Hakim G.: Stem cell factor and H2O2 induce GLUT1 translocation in M07e cells. Biofactors, 2004; 20: 97-108
[PubMed]
[46] Maschek G., Savaraj N., Priebe W., Braunschweiger P., Hamilton K., Tidmarsh G.F., De Young L.R., Lampidis T.J.: 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res., 2004; 64: 31-34
[PubMed] [Full Text HTML] [Full Text PDF]
[47] Maxwell P.H., Dachs G.U., Gleadle J.M., Nicholls L.G., Harris A.L., Stratford I.J., Hankinson O., Pugh C.W., Ratcliffe P.J.: Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA, 1997; 94: 8104-8109
[PubMed] [Full Text HTML] [Full Text PDF]
[48] Melstrom L.G., Salabat M.R., Ding X.Z., Milam B.M., Strouch M., Pelling J.C., Bentrem D.J.: Apigenin inhibits the GLUT-1 glucose transporter and the phosphoinositide 3-kinase/Akt pathway in human pancreatic cancer cells. Pancreas, 2008; 37: 426-431
[PubMed]
[49] Mitani T., Hoshikawa H., Mori T., Hosokawa T., Tsukamoto I., Yamaguchi F., Kamitori K., Tokuda M., Mori N.: Growth inhibition of head and neck carcinomas by D-allose. Head Neck, 2009; 31: 1049-1055
[PubMed]
[50] Moin S.M., Chandra V., Arya R., Jameel S.: The hepatitis E virus ORF3 protein stabilizes HIF-1α and enhances HIF-1 mediated transcriptional activity through p300/CBP. Cell. Microbiol., 2009; 11: 1409-1421
[PubMed]
[51] Mori Y., Tsukinoki K., Yasuda M., Miyazawa M., Kaneko A., Watanabe Y.: Glucose transporter type 1 expression are associated with poor prognosis in patients with salivary gland tumors. Oral Oncol., 2007; 43: 563-569
[PubMed]
[52] Murakami T., Nishiyama T., Shirotani T., Shinohara Y., Kan M., Ishii K., Kanai F., Nakazuru S., Ebina Y.: Identification of two enhancer elements in the gene encoding the type 1 glucose transporter from the mouse which are responsive to serum, growth factor and oncogenes. J. Biol. Chem., 1992; 267: 9300-9306
[PubMed] [Full Text PDF]
[53] Naftalin R.J.: Alternating carrier models of asymmetric glucose transport violate the energy conservation laws. Biophys J., 2008; 95: 4300-4314
[PubMed] [Full Text HTML] [Full Text PDF]
[54] Nakajo M., Kajiya Y., Tani A., Yoneda S., Shirahama H., Higashi M., Nakajo M.: 18FDG PET for grading malignancy in thymic epithelial tumors: significant differences in 18FDG uptake and expression of glucose transporter-1 and hexokinase II between low and high-risk tumors: Preliminary study. Eur. J. Radiol., 2012; 81: 146-151
[PubMed]
[55] Noguchi Y., Saito A., Miyagi Y., Yamanaka S., Marat D., Doi C., Yoshikawa T., Tsuburaya A., Ito T., Satoh S.: Suppression of facilitative glucose transporter 1 mRNA can suppress tumor growth. Cancer Lett., 2000; 154: 175-182
[PubMed]
[56] Noto Y., Iwazaki A., Nagao J., Sumiyama Y., Redpath J.L., Stanbridge E.J., Kitagawa T.: Altered N-Glycosylation of glucose transporter-1 associated with radiation-induced tumorigenesis of human cell hybrids. Biochem. Biophys. Res. Commun., 1997; 240: 395-398
[PubMed]
[57] Nurzyńska D.: Hamowanie onkogenów za pomocą oligonukleotydów antysensowych jako metoda terapii nowotworów. Współczesna Onkologia, 2003; 7: 18-23
[Abstract]
[58] Osthus R.C., Shim H., Kim S., Li Q., Reddy R., Mukherjee M., Xu Y., Wonsey D., Lee L.A., Dang C.V.: Deregulation of glucose transporter 1 and glycolytic gene expression by c-myc. J. Biol. Chem., 2000; 275: 21797-21800
[PubMed] [Full Text HTML] [Full Text PDF]
[59] Pagé E.L., Chan D.A., Giaccia A.J., Levine M., Richard D.E.: Hypoxia-inducible factor-1α stabilization in nonhypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol. Biol. Cell, 2008; 19: 86-94
[PubMed] [Full Text HTML] [Full Text PDF]
[60] Pérez A., Ojeda P., Valenzuela X., Ortega M., Sánchez C., Ojeda L., Castro M., Cárcamo J.G., Rauch M.C., Concha I.I., Rivas C.I., Vera J.C., Reyes A.M.: Endofacial competitive inhibition of the glucose transporter 1 activity by gossypol. Am. J. Physiol. Cell Physiol., 2009; 297: c86-c93
[PubMed] [Full Text HTML] [Full Text PDF]
[61] Ravazoula P., Batistatou A., Aletra C., Ladopoulos J., Kourounis G., Tzigounis B.: Immunohistochemical expression of glucose transporter Glut1 and cyclin D1 in breast carcinomas with negative lymph nodes. Eur. J. Gynaecol. Oncol., 2003; 24: 544-546
[PubMed]
[62] Rees W.D., Holman G.D.: Hydrogen bonding requirements for the insulin-sensitive sugar transport system of rat adipocytes. Biochim Biophys. Acta, 1981; 646: 251-260
[PubMed]
[63] Robichaud T., Appleyard A.N., Herbert R.B., Henderson P.J., Carruthers A.: Determinants of ligand binding affinity and cooperativity at the GLUT1 endofacial side. Biochemistry, 2011; 50: 3137-3148
[PubMed]
[64] Rosner M., Hengstschläger M.: Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum. Mol. Genet., 2008; 17: 2934-2948
[PubMed] [Full Text HTML] [Full Text PDF]
[65] Roux P.P., Ballif B.A., Anjum R., Gygi S.P., Blenis J.: Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA, 2004; 101: 13489-13494
[PubMed] [Full Text HTML] [Full Text PDF]
[66] Samih N., Hovsepian S., Notel F., Prorok M., Zattara-Cannoni H., Mathieu S., Lombardo D., Fayet G., El-Battari A.: The impact of N– and O-glycosylation on the functions of Glut-1 transporter in human thyroid anaplastic cells. Biochim. Biophys. Acta, 2003; 1621: 92-101
[PubMed]
[67] Santalucía T., Christmann M., Yacoub M.H., Brand N.J.: Hypertrophic agonists induce the binding of c-Fos to an AP-1 site in cardiac myocytes: implications for the expression of GLUT1. Cardiovasc. Res., 2003; 59: 639-648
[PubMed] [Full Text HTML] [Full Text PDF]
[68] Sato M., Mueckler M.: A conserved amino acid motif (R-X-G-R-R) in the Glut1 glucose transporter is an important determinant of membrane topology. J. Biol. Chem., 1999; 274: 24721-24725
[PubMed] [Full Text HTML] [Full Text PDF]
[69] Saxena N., Lahiri S.S., Hambarde S., Tripathi R.P.: RAS: target for cancer therapy. Cancer Invest., 2008; 26: 948-955
[PubMed]
[70] Schwartzenberg-Bar-Yoseph F., Armoni M., Karnieli E.: The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res., 2004; 64: 2627-2633
[PubMed] [Full Text HTML] [Full Text PDF]
[71] Seatter M.J., De la Rue S.A., Porter L.M., Gould G.W.: QLS motif in transmembrane helix VII of the glucose transporter family interacts with the C-1 position of D-glucose and is involved in substrate selection at the exofacial binding site. Biochemistry, 1998; 37: 1322-1326
[PubMed]
[72] Semenza G.L.: Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J., 2007; 405: 1-9
[PubMed] [Full Text HTML] [Full Text PDF]
[73] Sergeant S. Kim H.D.: Inhibition of 3-O-methylglucose transport in human erythrocytes by forskolin. J. Biol. Chem., 1985; 260: 14677-14682
[PubMed] [Full Text PDF]
[74] Shafee N., Kaluz S., Ru N., Stanbridge E.J.: PI3K/Akt activity has variable cell-specific effects on expression of HIF target genes, CA9 and VEGF, in human cancer cell lines. Cancer Lett., 2009; 282: 109-115
[PubMed] [Full Text HTML] [Full Text PDF]
[75] Shahbazian D., Roux P.P., Mieulet V., Cohen M.S., Raught B., Taunton J., Hershey J.W., Blenis J., Pende M., Sonenberg N.: The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J., 2006; 25: 2781-2791
[PubMed] [Full Text HTML] [Full Text PDF]
[76] Shim H., Dolde C., Lewis B.C., Wu C.S., Dang G., Jungmann R.A., Dalla-Favera R., Dang C.V.: c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA, 1997; 94: 6658-6663
[PubMed] [Full Text HTML] [Full Text PDF]
[77] Silva A., Gírio A., Cebola I., Santos C.I., Antunes F., Barata J.T.: Intracellular reactive oxygen species are essentials for PI3K/Akt/mTOR-dependent IL-7 mediated viability of T-cell acute lymphoblastic leukemia cells., Leukemia, 2011; 25: 960-967
[PubMed]
[78] Singh D., Banerji A.K., Dwarakanath B.S., Tripathi R.P., Gupta J.P., Mathew T.L., Ravindranath T., Jain V.: Optimizing cancer radiotherapy with 2-deoxy-D-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther. Oncol., 2005;181: 507-514
[PubMed]
[79] Sparks C.A., Guertin D.A.: Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene, 2010; 29: 3733-3744
[PubMed] [Full Text HTML] [Full Text PDF]
[80] Steelman L.S., Chappell W.H., Abrams S.L., Kempf C.R., Long J., Laidler P., Mijatovic S., Maksimovic-Ivanic D., Stivala F., Mazzarino M.C., Donia M., Fagone P., Malaponte G., Nicoletti F., Libra M., Milella M., Tafuri A., Bonati A., Bäsecke J., Cocco L., Evangelisti C., Martelli A.M., Montalto G., Cervello M., McCubrey J.A.: Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging, 2011; 3: 192-222
[PubMed] [Full Text HTML] [Full Text PDF]
[81] Subbarayan P.R., Wang P.G., Lampidis T.J., Ardalan B., Braunschweiger P.: Differential expression of GLUT1 mRNA and protein levels correlates with increased sensitivity to the glyco-conjugated nitric oxide donor (2-glu-SNAP) in different tumor cell types. J. Chemother., 2008; 20: 106-111
[PubMed]
[82] Sui L., Dong Y., Watanabe Y., Yamaguchi F., Hatano N., Tsukamoto I., Izumori K., Tokuda M.: The inhibitory effect and possibile mechanisms of D-allose on cancer cell proliferation. Int. J. Oncol., 2005; 27: 907-912
[PubMed]
[83] Sun Y., Fang Y., Yoon M.S., Zhang C., Roccio M., Zwartkruis F.J., Armstrong M., Brown H.A., Chen J.: Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc. Natl. Acad. Sci. USA, 2008; 105: 8286-8291
[PubMed] [Full Text HTML] [Full Text PDF]
[84] Thangaraju M., Carswell K.N., Prasad P.D., Ganapathy V.: Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem. J., 2009; 417: 379-389
[PubMed] [Full Text HTML] [Full Text PDF]
[85] Thorens B., Mueckler M.: Glucose transporters in the 21st century. Am. J. Physiol. Endocrinol. Metab., 2010; 298: E141-E145
[PubMed] [Full Text HTML] [Full Text PDF]
[86] Uhlenbrock K., Weiwad M., Wetzker R., Fischer G., Wittinghofer A., Rubio I.: Reassessment of the role of FKBP38 in the Rheb/mTORC1 pathway. FEBS Lett., 2009; 583: 965-970
[PubMed]
[87] Uldry M., Ibberson M., Hosokawa M., Thorens B.: GLUT2 is a high affinity glucosamine transporter. FEBS Lett., 2002; 524: 199-203
[PubMed]
[88] Usuda K., Sagawa M., Aikawa H., Ueno M., Tanaka M., Machida Y., Zhao X.T., Ueda Y., Higashi K., Sakuma T.: Correlation between glucose transporter-1 expression and 18F-fluoro-2-deoxyglucose uptake on positron emission tomography In lung cancer. Gen. Thorac. Cardiovasc. Surg., 2010; 58: 405-410
[PubMed]
[89] Varshney R., Dwarakanath B.S., Jain V.: Radiosensitization by 6-aminonicotinamide and 2-deoxy-D-glucose in human cancer cells. Int. J. Radiat. Biol., 2005; 81: 397-408
[PubMed]
[90] Vera J.C., Reyes A.M., Cárcamo J.G., Velásquez F.V., Rivas C.I., Zhang R.H., Strobel P., Iribarren R., Scher H.I., Slebe J.C., Golde D.W.: Genistein is a natural inhibitor of hexose and dehydroascorbic acid transport through the glucose transporter, GLUT1. J. Biol. Chem., 1996; 271: 8719-8724
[PubMed] [Full Text HTML] [Full Text PDF]
[91] Vera J.C., Rosen O.M.: Functional expression of mammalian glucose transporters in Xenopus leavis oocytes: evidence for cell-dependent insulin sensitivity. Mol. Cell. Biol., 1989; 9: 4187-4195
[PubMed] [Full Text HTML] [Full Text PDF]
[92] Wang L., Harris T.E., Lawrence J.C.Jr: Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J. Biol. Chem., 2008; 283: 15619-15627
[PubMed] [Full Text HTML] [Full Text PDF]
[93] Watanabe R., Wei L., Huang J.: mTOR signaling, function, novel inhibitors, and therapeutic targets. J. Nucl. Med., 2011; 52: 497-500
[PubMed]
[94] Wieman H.L., Horn S.R., Jacobs S.R., Altman B.J., Kornbluth S., Rathmell J.C.: An essential role for the Glut1 PDZ-binding motif in growth factor regulation of Glut1 degradation and trafficking. Biochem J., 2009; 418: 345-367
[PubMed] [Full Text HTML] [Full Text PDF]
[95] Wood I.S., Trayhurn P.: Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br. J. Nutr., 2003; 89: 3-9
[PubMed]
[96] Xie H., Valera V.A., Merino M.J., Amato A.M., Signoretti S., Linehan W.M., Sukhatme V.P., Seth P.: LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol. Cancer Ther., 2009; 8: 626-635
[PubMed] [Full Text HTML] [Full Text PDF]
[97] Yun J., Rago C., Cheong I., Pagliarini R., Angenendt P., Rajagopalan H., Schmidt K., Willson J.K., Markowitz S., Zhou S., Diaz L.A.Jr, Velculescu V.E., Lengauer C., Kinzler K.W., Vogelstein B., Papadopoulos N.: Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science, 2009; 325: 1555-1559
[PubMed] [Full Text HTML] [Full Text PDF]
[98] Zhao F.Q., Keating A.F.: Functional properties and genomics of glucose transporters. Curr. Genomics, 2007; 8: 113-128
[PubMed] [Full Text HTML] [Full Text PDF]
[99] Zhou H., Huang S.: mTOR signaling in cancer cell motility and tumor metastasis. Crit. Rev. Eukaryot. Gene Expr., 2010; 20: 1-16
[PubMed] [Full Text HTML] [Full Text PDF]
[100] Zottola R.J., Cloherty E.K., Coderre P.E., Hansen A., Hebert D.N., Carruthers A.: Glucose transporter function is controlled by transporter oligomeric structure. A single, intramolecular disulfide promotes GLUT1 tetramerization. Biochemistry, 1995; 34: 9734-9747
[PubMed]
Autorzy deklarują brak potencjalnych konfliktów interesów.