
Tyrosine kinase inhibitors in hematological malignancies
Kamila Kosior 1 , Magdalena Lewandowska-Grygiel 2 , Krzysztof Giannopoulos 3Streszczenie
W ostatnich latach nowe strategie leczenia nowotworów opierają się głównie na terapii celowanej. Kinazy tyrozynowe ze względu na to, iż przenoszą reszty fosforanowe z adenozynotrifosforanu (ATP) na tyrozynę uczestnicząc w transmisji najważniejszych sygnałów międzykomórkowych niezbędnych w procesach wzrostowych, związanych między innymi z kancerogenezą, stanowią dobry cel w terapii przeciwnowotworowej. Zaburzenia w regulacji aktywności kinaz tyrozynowych są często stwierdzane w rozrostowych chorobach hematologicznych, szczególnie obserwowano nadekspresję kinazy tyrozynowej w przewlekłej białaczce szpikowej oraz ostrej białaczce limfoblastycznej. W pracy opisano znaczenie inhibitorów kinazy tyrozynowej w hematoonkologii. Leki te zrewolucjonizowały terapię przeciwbiałaczkową i są jednymi z najbardziej aktywnych związków terapeutycznych dających nadzieję nie tylko na osiągnięcie długotrwałej remisji, ale także na całkowite wyleczenie. Publikacja podsumowuje aktualną wiedzę dotyczącą imatynibu – inhibitora kinazy tyrozynowej pierwszej generacji, dasatynibu i nilotynibu – inhibitorów kinazy tyrozynowej drugiej generacji oraz bosutynibu – inhibitora kinazy tyrozynowej trzeciej generacji, jak również nowych inhibitorów kinazy tyrozynowej ponatynibu i danusertibu.
Słowa kluczowe:inhibitory kinazy tyrozynowej (TKI) • przewlekła białaczka szpikowa (CML) • ostra białaczka limfo blastyczna (ALL) • imatinib • dasatinib • nilotinib • bosutinib
Summary
Recently novel treatment modalities has focused on targeted therapies. Tyrosine kinases represent a good target for cancer treatment since they are involved in transferring phosphate groups from ATP to tyrosine residues in specific substrate proteins transducing intracellular signals engaged in the many mechanisms, playing an important role in the modulation of growth factors signaling that are strongly related to carcinogenesis. Deregulation of tyrosine kinases activity was also found in hematological malignancies, particularly overexpression of tyrosine kinases was observed in chronic myeloid leukemia or acute lymphoblastic leukemia. Herein we show that tyrosine kinase inhibitors have revolutionized hematology malignancies therapy in a very short period of time and they still remain one of the most interesting anticancer compounds that could give a hope for cure and not only long-lasting complete remission. This manuscript summarizes current view on the first generation tyrosine kinase inhibititor – imatinib, second generation – dasatinib, nilotinib and bosutnib as well as new generation tyrosine kinase inhibititors – ponatinib and danusertib in hematooncology.
Key words:tyrosine kinase inhibitors (TKI) • chronic myeloid leukemia (CML) • acute lymphoblastic leukemia (ALL) • imatinib • dasatinib • nilotinib • bosutinib
Introduction
Despite better understanding of the patomechanism of hematological malignancies and novel therapeutic strategies most of leukemias remains incurable. Conventional chemotherapy or bone marrow transplantation, only in some cases, is able to completely eliminate the disease. Moreover, most therapies are accompanied with adverse effects that could even limit patient’s survival in certain circumstances. Therefore there is a growing interest in targeted therapies in recent years. These treatments are also often associated with lower toxicity and better direct action against cancer-specific molecules or signaling pathways which are specifically deregulated in tumorigenesis [3]. It has been proved that tyrosine kinases (TK) represent a good target for cancer therapy. They are involved in the most important mechanisms controlling cell growth and proliferation. They play an important role in the modulation of growth factor signaling which is strongly related to carcinogenesis, participating in various cellular processes including cell growth, proliferation, differentiation, and cell death. Tyrosine kinases are subclass of substrate protein kinases involved in transferring phosphate groups from ATP to tyrosine residues in specific substrate proteins transducting intracellular signals (fig. 1). Some changes caused by gene mutation can lead to overproduction of proteins functioning as tyrosine kinases, which are responsible for proliferation of particular cell lines, what results in uncontrolled cell proliferation and resistance to apoptotic stimuli.

Figure 1. Mechanism of action of tyrosine kinase. Tyrosine kinase catalyzes the transfer of a phosphate group from ATP to the side chains of tyrosine residues, altering inactive into active phosphorylated conformation of protein
Deregulation of TK activity contributes to development of neoplastic diseases, therefore controlling of their activity can have a principal meaning in the treatment. Oncogenic TK, which are expressed in malignant tumors, inhibit apoptosis, alter cell adhesion and stimulate growth factor-independent cell proliferation. It was found that oncogenic TK exhibit increased enzymatic activity compared with the proteins of normal cells [16,39,85].
Modulation of tyrosine kinase activity in chronic myeloid leukemia and acute lymphoblastic leukemia
The most common aberrantly activated TK is protein BCR/ABL (p210), which is a product of gene associated with Philadelphia chromosome. This unique chromosome is generated by translocation t(9,22)(q34: q11) which produces aberrant TK BCR/ABL (fig. 2). This mutation is present virtually in most cases of CML (>95%). Chronic myeloid leukemia (CML) is a hematologic malignancy, where reciprocal translocation involving the long arms of chromosomes 9 and 22, resulting in the BCR/ABL fusion gene, plays a crucial role of its pathogenesis and a clinical course. Although occurrence of fusion protein (p210) probably is not a sole factor responsible for pathogenesis of this type of leukemia and it is not present in all patients, it still represents the best target for CML therapy [2,4,12,41,52,64,72,91]. Earlier cytoreductive treatment followed by interferon alpha (IFN-α) as well as combined chemotherapy was used as the main treatment strategy for CML. Introduction of IFN-α which is nonspecific immunostimulant that regulates T-cell activity improved survival of CML patients but therapy was rather weakly tolerated. In many patients flu-like symptoms, depression and mood changes were observed. In addition this quite toxic therapy has required 3 injections per week, which was inconvenient and quite expensive for many patients [19].These facts prompted the exploration of new drugs for the treatment for CML more specific and better tolerated.
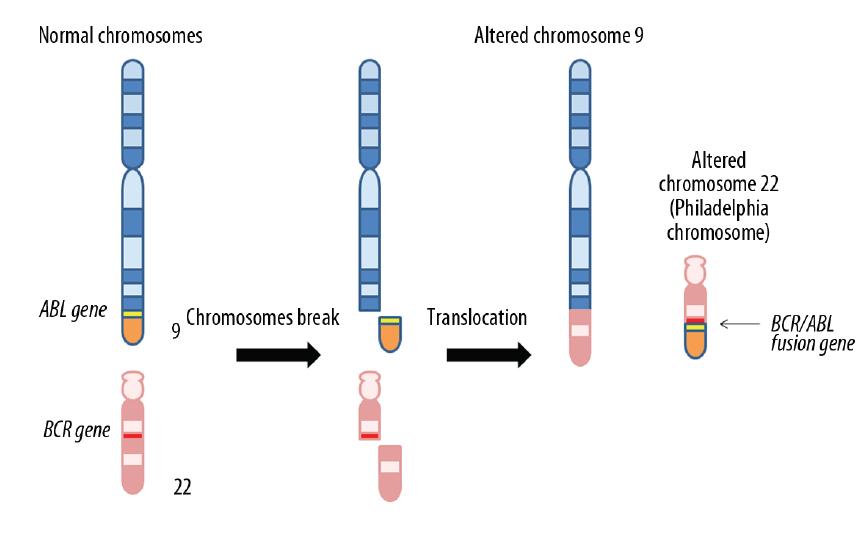
Figure 2. Fusion of chromosomes 9 and 22 results in generation of the Philadelphia chromosome. Philadelphia chromosome is generated by translocation t(9,22)(q34: q11) which produces aberrant tyrosine kinase BCR/ABL. This mutation is present virtually in most cases of CML (>95%)
For the facts mentioned above, TKI have seemed to be interesting agents, which could block proliferation of CML cells and lead to their apoptosis. Since TKI act directly on enzymes engaged in neoplastic process, they appear to be a good candidates used in certain types of cancer, especially, in disorders driven by overexpression of TK like the protein product of BCR-ABL fusion gene in CML.
Imatinib mesylate
Imatinib mesylate, as the first TKI, has revolutionized the approach of CML treatment and set a new standard in CML patients’ therapy. A complete cytogenetic response was achieved in 50% to 60% of patients treated in chronic phase after failure with interferon therapy and in over 80% of newly diagnosed patients [43]. Imatinib mesylate, originally STI571 (Signal Transduction Inhibitor 571), has been commercially available since May 2001 and since then has been used as a first-line agent for newly diagnosed patients in CML [19,80,92]. Recently also dasatinib and nilotinib – second kinase inhibitors – were approved as agents for first line treatment in patients with chronic phase of CML [87]. Imatinib, besides being better tolerated than INF-α, is also much more potent in prolonging progression-free survival and in reducing BCR/ABL transcript levels in CML [92]. Imatinib was the first agent found to be a clinically effective by specific inhibition of genetic aberration which plays a role in a malignant transformation. Its mechanism of action bases on blocking the ATP binding to the inactive BCR/ABL conformation, preventing thereby tyrosine phosphorylation on proteins responsible for intracellular signal transduction, what leads to inhibition of proliferation and leukemia cells apoptosis. Furthermore, recent studies revealed that imatinib inhibits also PDGFR (receptor for platelet-derived growth factor) and c-Kit (receptor for the cytokine stem cell factor), not solely Abl kinases [9,32,80].
Clinically CML involves three main phases: chronic, accelerated and blast crisis. The latter, most advanced, is similar to acute leukemia. Occasionally, also transformation to ALL appears [29]. Translocation (9;22) is also the most frequent genetic rearrangement in ALL therefore imatinib appeared to be a good agent for treatment of relapsed and refractory Ph+ ALL patients, when convectional chemotherapy and stem cell transplantation failed [65]. During progression of the ALL proliferation and accumulation of immature blast cells results in suppression of normal haematopoiesis [62]. Imatinib revealed to be highly effective in patients with Ph+ ALL [86]. Unfortunately, imatinib used as a sole treatment in Ph+ ALL induced a short responses with median time to progression over 2 months and overall survival approximately 5 months. It prompted further studies on mechanism of resistance development to imatinib alone or in combination with other agents [59]. As it turned out, also in a relatively short time after completing clinical trials on imatinib in CML, in some patients, resistance and intolerance to imatinib was observed. From that time, the mechanism of resistance to imatinib has been extensively studied. It was noticed that resistance more common appears in accelerated phase or blast crisis, even after complete hematological response [74,82]. Underlying mechanisms of resistance have been divided into two main groups: BCR/ABL-dependent and -independent. The first, ABL-kinase domain point mutation could change sensitivity to imatinib in BCR/ABL transformed cells. Presence of such mutation leads to lower sensitivity to TKI via alteration of amino acids residues in inhibitor binding site or preventing enzyme from achieving inactive conformation required for imatinib binding [35,76]. Up till now more than 50 different mutants have been described, some of them are characteristic for chronic phase, accelerated or blast crisis. The scientists observed, that most of these mutations are intimately concerned with amino acids substitution at seven residues (M244V, G250E, Y253F/H, E255K/V, T315I, M351T and F359V). It was also demonstrated, that severity of disease has an impact on occurrence of resistance-associated mutations. CML patients in advanced phases and Ph+ ALL patients more often develop mutation. Especially presence of mutation at P-loop (glycine-rich sequence spanning amino-acids from 248 to 256, which binds with imatinib through hydrogen and van der Waals bonds) is related to decreased sensitivity to imatinib via destabilization of conformation required for imatinib biding. In particular, complete resistance to imatinib was noticed in patients with T315I mutation described by Gorre et al. in 2001 [27], and was also considered as a bad prognostic factor. Thereby presence of mentioned mutation possesses the major warning that therapeutic strategy should be reconsidered [7,79]. The second usually reported reason of imatinib resistance concerning BCR-ABL gene is its uncontrolled amplification, however its more pronounced in in vitro studies than in clinic [27,34,73].
Other TK are also essential for signal transduction required to control proliferation and apoptosis. Overexpression of Lyn kinase (member of the Src family of protein tyrosine kinases, which is mainly expressed in hematopoetic cells) appears to play an important role in the survival of CML cells that are resistant to imatinib. Studies of Donato et al. [15] on CML cell line K562R have shown that Lyn kinase is responsible for sensitivity of CML cells to the inhibition of BCR/ABL kinase, and that there are differences in Lyn regulation between imatinib-sensitive and imatinib-resistant CML cell lines. Further studies also confirm this finding [61,93].
Other factors associated with enhanced proliferation represents BCR/ABL independent cytogenetic aberrations. The main cause for imatinib resistance in this group of patients is a clonal evolution with presence of additional cytogenetic aberration. Hochhaus et al. [34] studies revealed that novel cytogenetic aberrations could be detected in 19 of 36 with refractory CML patients. In 13 aneuploidy association with chromosomal instability was found. Also the loss of one p53 allele, caused by changes in the short arm of chromosome 17 occurred in seven patients [17].
Other groups also studied an impact of P-glycoprotein (P-gp ABCB-1) overexpression on imatinib resistance, since imatinib is effectively effluxed by P-gp. This integral membrane protein is one of members of ATP-binding cassette (ABC) transporters, which is composed of two homologous parts, each containing the site for binding cytosolic ATP. The energy obtained from ATP-hydrolysis is used to transport P-gp substrates and keep them out of cells diminishing their intracellular concentration [37,38]. The studies showed the correlation between accumulation of P-gp protein and worse response to imatinib also the severity of the disease was dependent on P-gp expression. Since P-gp is a product of multi-drug resistance gene – MDR1, some investigations found polymorphism of MDR1 revealed molecular resistance to imatinib [13,40].
Other important factor involved in imatinib cellular distribution is h-OTC-1 protein (human organic cation transporter-1). Since imatinib is moved into cell via mechanism of active transport by h-OTC-1, impaired function could also lead to imatinib resistance. This hypothesis faced by White et al. [89], where authors proved that patients with low expression of this protein exhibit suboptimal response to imatinib. In addition, they used various doses of imatinib and observed that patients who express low levels of h-OTC-1 and receive higher doses (more than 600 mg) of imatinib responded better to therapy. Moreover, imatinib plasma concentration was assayed in patients with good responses and compared with those with imatinib resistance. The obtained results suggest relation between response to imatinib and its plasma concentration. This fact is probably associated with strong binding of imatinib to alpha1-acid glycoprotein [48].
Specific inhibition of TK was also found as a therapeutic strategy in gastrointestinal stromal tumors (GISTs) [47].
Second generation tyrosine kinase inhibitors
Although imatinib has proved efficacy in a first-line therapy for CML and Ph+ ALL patients, above mentioned observations on resistance prompted further studies in the quest for new agents among TKI which could produce a response in patients with imatinib-resistant or refractory CML or Ph+ALL. Novel compounds of this group of drugs, represent second generation TKI, which turned out to be a good option for CML patients who do not tolerate imatinib, harbor the mutation or exhibit imatinib resistance from another reasons.
Dasatinib
Dasatinib (formerly BMS354825) is a multitarget TKI effective in the majority of imatinib resistance leukemias. It blocks activity of BCR/ABL and all members of Src-family kinases. Like imatinib, it has also a capability to block other kinases such as c-KIT, PDGFR and ephrin receptor kinase [58]. Initially, since June 2006 dasatinib has been used for imatinib-resistant or intolerant CML and Ph+ ALL and since October 2010 it has been approved in 100 mg once a day as a first-line treatment of chronic phase for newly diagnosed CML.
Like imatinib, it is an ATP-competitive kinase inhibitor but it has a different chemical structure [50]. As it was mentioned, some of mutations causing resistance to imatinib were involved in changing kinase conformation, making it unavailable to this compound. Dasatinib seems to be more effective than imatinib because of its mechanism of binding to the ABL kinase domain, which doesn’t require an inactive conformation. It can bind to both the inactive and active forms of BCR/ABL. Thanks that dasatinib can overcome almost all imatinib-resistant forms of Bcr-Abl. Only the T315I mutant remains resistant also to this second generation TKI. Moreover, the in vitro studies proved that dasatinib is over 300-fold more potent against wild-type BCR/ABL than imatinib [56,58,77,83]. As mentioned above, clinical trials revealed that unsatisfactory results after treatment with imatinib in some patients with CML may be also related to overexpression of Src kinases, especially Hck and Lyn [90]. Taking under consideration that dasatinib is also active against this family of kinases, it appears to be a perfect compound to overcome this source of imatinib resistance [15,61]. Konig at al. [45] revealed that Src phosphorylation is also enhanced in progenitor cells and it is involved in leukemogenesis in CML. This is another resistance to imatinib mechanism thereby dasatinib in regard of targeting on Src signaling pathway could additionally target on progenitors cells in CML. It was proved that dasatinib reduced Src level in CD34+ and in more primitive CD34+CD38- cells in all phases of CML. Studies also showed that dasatinib inhibits both BCR/ABL kinase-dependent and kinase-independent Src activation, while imatinib acts solely on BCR/ABL kinase mediated Src activation. In experiment on CML CD34+ cells exposed to imatinib (5 Amol/L) or dasatinib (0.01-0.15 Amol/L) with or without exogenous growth factor (GF) authors found that dasatinib in the absence of GF was able to reduce MAPK, Akt, and Stat5 (signal transducer and activator of transcription 5) expression [15,93]. Stat proteins and its downstream transducers play a crucial role in many physiopathological processes, including controlling of immune response and cell cycle. Stat5 is the most important protein of Stat family. Deregulation of Stat5 signaling pathways is related to immunosuppression, enhanced proliferation and survival of pathological cells. Hoelbl et al. [36] has observed that Stat5 is crucial for viability and proliferation of leukaemic cells in Ph-positive CML and ALL. They proved that inhibition of Stat5 led to G0/G1 cell cycle arrest and apoptosis of leukemic cells in vitro and elimination of myeloid and lymphoid leukemia cells in vivo. Nam et al. [55] observed that dasatinib has an ability to decrease levels of phosphorylated Stat5 and Stat5-DNA binding activity. Authors further observed reduced expression of some Stat5 targets such as Bcl-x, Mcl-1 and cyclin D1 which are related to cancer progression. Hence, inhibition of Stat5 – as well as alone or in combination with BCR/ABL – may pose a new effective strategy toward treatment of CML.
As has been observed in many studies that Src kinases, especially Lyn, which has the greatest expression in myeloid cells, play an important role in myelopoiesis. Hence, inhibition of its activity appears to be a good therapy of acute myeloid leukemia (AML). Interestingly, to prove that silencing of Lyn gives good result in arrestment of myeloid cell growth, the scientists used interference with siRNA to decrease level of Lyn kinases. Nonetheless, RNA interference has not been used yet in therapeutic regimens, thus they have continued this study with dasatinib as the Src kinase inhibitor. After 60-minute incubation with dasatinib at nanomolar concentrations, the inhibition was observed in all of studied cell lines. The scientists used in this study most characteristic cell lines for AML. The result of some other experiments have revealed that dasatinib potently inhibited the growth of primary AML blasts.
Dasatinib efficacy against c-Kit augments its further utilization in AML. Guerrouahen et al. [30] used Mo7e cells line, which harbor an activating mutation of Kit and treated with dasatinib. They observed direct inhibition of c-Kit and effective inhibition of the growth of Mo7e cells [36,53,94].
Another important factor affecting effectiveness of therapy is mechanism of drug cellular transport. It has been also mentioned that expression of ATP binding-cassette member – ABCB-1 – can influence on imatinib intracellular concentration and thereby limiting its efficacy in CML patients. In contrast to imatinib, dasatinib has not been a substrate of P-gp, therefore it appeared to be more effective than imatinib due to higher intracellular concentration in hematopoietic progenitor cells [81]. Surprisingly, the recent results revealed that dasatinib has also an affinity for dissolving in lipids and could also be a substrate of ABCB-1. Hiwase et al. [33] also confirmed that dasatinib is also a substrate of ABCB-2 efflux transporter. Other protein involved in cellular drug transport is h-OTC-1. As has been shown imatinib uptake is dependent on this active protein, which function depends on temperature. Studies on dasatinib indicated no difference between dasatinib concentrations in 4°C and 37°C, what suggests that dasatinib uptake is a more passive process. Also inhibition of h-OCT-1 did not change dasatinib concentration, in contrast to imatinib. These data proved that dasatinib uptake does not depend on h-OCT-1 level. Therefore treatment with dasatinib may be a good strategy for patients with low h-OCT-1 expression [24,42]. Above findings suggest that characterization of the cellular uptake and efflux pathways of the TKI may help to define appropriate and effective therapy for individual CML patients.
Studies on dasatinib therapy for newly diagnosed CML patients in comparison with the treatment with standard dose of imatinib has shown, that patients with chronic-phase treated with dasatinib, in 100 mg/day, achieved a complete cytogenetic response and a major molecular response to a greater extent than those treated with imatinib 400 mg/day. Additionally, dasatinib induced responses faster and more potently than imatinib, therefore on October 28, 2010, it was approved by the US Food and Drug Administration at 100 mg once daily as an agent used in the treatment for patients with a newly diagnosed chronic phase and at 70 mg twice daily in patients with more advanced phases of CML [44].
It has also been proved that dasatinib is effective in patient with Ph-positive ALL. Recent studies have shown a big efficacy of dasatinib in untreated patient with Ph-positive ALL. Foa et al. [22] have revealed that all of patients with newly diagnosed Ph-positive ALL who were treated with dasatinib, combined with steroids, achieved complete response within one month on treatment. Also in Ravandi’s study [68] has shown that dasatinib in combination with hyperCVAD caused complete response in 33 (94%) patients. These findings indicate that dasatinib as a first-line treatment in Ph-positive ALL, combined with steroids and chemotherapy, appears to be a good therapeutic option [68]. Immunomodulatory character of dasatinib appears to be very controversial. Some studies have suggested that dasatinib, in regard of targeting Src kinases, may have an suppressive character. In 2007 Schade et al. [75] have shown, that dasatinib can inhibit T-cell activation and proliferation via inhibition of LCK, which is crucial for T-cell receptor (TCR) signaling pathway. They observed inhibition of TCR-mediated signal transduction, cellular proliferation, cytokine production, and in vivo T-cell responses. This finding encouraged further studies on immunomodulatory proprieties of dasatinib. Another aspect of immunosuppression is inhibition of NK cells cytotoxicity as some Src kinases are essential for maintenance of this NK function. The results of the study [6] revealed that dasatinib reduced NK cell cytotoxicity with maximal effect achieved at about 25 nM. But there has not been proved in in vivo studies yet. Perhaps in the case of T-cells only high doses of dasatinib have a suppressive properties. This findings, especially associated with suppression of T-cell activity appears to be helpful in therapy for patients with molecular relapse in CML after haploidentical BMT with chronic GVHD (graft-versus-host disease). Breccia et al. [8] carried the investigation on patients, who relapsed and had chronic liver GVHD after BMT. They have shown that dasatinib at low doses was well-tolerated and restored the molecular response and improved the hepatic dysfunction caused by GVHD. Interestingly, the recent publications have shown, that dasatinib influences on increase of lymphocytosis in patient with Ph positive leukemia. In 2010, Kreutzman and colleagues [46] observed the presence of clonal cytotoxic T and NK cells in over 80% (15 of 18) of patients with CML during imatinib therapy. What is more interesting, they described increase of the populations after further treatment with dasatinib. Earlier Mustjoki et al. [54] have shown increase of NK/T cells in 22 patients with CML and Ph+ ALL treated with dasatinib. He also demonstrated correlation between the development of lyphocytosis and better effects of therapy. Mechanism of this expansion has not been explained yet, but comparable results have been observed by others. Interestingly, only patients who developed lymphocytosis demonstrated higher rates of cumulative complete cytogenetic response and major cytogenetic response at one year [8,10,67,75].
Nilotinib
Other second-generation TKI – nilotinib – originally termed AMNI07, like dasatinib, has been initially used in the treatment for adults in chronic and accelerated phase of Ph-positive CML resistant or intolerant to imatinib. In June 2010 nilotinib received FDA approval as a first-line therapy in patients with chronic phase of CML. Nilotinib was designed based on detailed molecular studies on imatinib’s structure. It is improved derivative of imatinib and, like imatinib, also bind to the inactive conformation of Abl. It has a similar chemical structure, but demonstrates better topographical fit for the ABL protein thanks to modification of the methylpiperazinyl group of imatinib, and it is approximately 30-fold more effective than imatinib as it was shown in cell lines expressing BCR/ABL (fig. 3) [70,88]. Data from Weisberg et al. [88] suggest that nilotinib selectively inhibits BCR/ABL, Kit, and PDGFR tyrosine kinases, but does not influences any of the kinases required for IL-3 signaling, including JAK2. Nilotinib, in contrast to dasatinib, has minimal effect on Src family kinases, what has been associated with nilotinib resistance in vitro in samples with increased expression of Lyn kinase. Since Lyn is involved in erythropoiesis and Hck is essential for the survival of myeloid cells and B lymphocytes, it has been proposed that nilotinib may have a more safe side effect profile especially with regard to myelosuppression than dasatinib [14,49,51]. >From October 29, 2007 nilotinib 400 mg twice daily has been approved in the US and Europe for patients in chronic and accelerated phases of CML who are resistant or intolerant to imatinib. After further trials, imatinib and nilotinib in newly diagnosed CML patients, nilotinib 300 mg twice a day was approved in the US for previously untreated patients with chronic phase of CML [88].

Figure 3. Chemical structures of imatinib, dasatinib and nilotinib and their localization in tyrosine kinase BCR/ABL; A – Chemical structure of the first generation tyrosine kinase inhibitor – imatinib and its localization in tyrosine kinase BCR/ABL, B – Chemical structures of the second generation tyrosine kinase inhibitors – dasatinib and nilotinib and their localization in tyrosine kinase BCR/ABL
Laboratory data on nilotinib have revealed that inhibition of cell growth by nilotinib was associated with induction of apoptosis [88]. Similarly to dasatinib, has activity against most of the mutated forms of BCR/ABL kinase. However, despite grater efficacy, resistance still occurs including Y253H, E255K/V, F359C/V and T315I mutants. This patients, treated with nilotinib after imatinib failure, had significantly shorter progression-free survival. Also in vitro studies have revealed lower sensitivity of these mutated clones to nilotinib [69,88]. Since several studies underlined the probable role of Bim expression during imatinib-induced apoptosis, nilotinib also could act via regulation of Bim levels, that results in CML cell death [1,18]. Belloc and colleagues [5] experimentally verified whether Bim accumulation was linked to BCR/ABL tyrosine kinase activity. They inhibit BCR/ABL autophosphorylation using imatinib and nilotinib. Both agents inhibited 80% of ABL and BCR/ABL autophosphorylation. This inhibition was accompanied by a two fold increase in relative BimEL expression. Accumulation of Bim was followed by mitochondrial apoptosis. As far as impact on T regulatory cells is concerned, comparing results of studies on these three kinase inhibitors used in front-line CML therapy, we can observe that imatinib inhibits the proliferation and function of Tregs and CD8+ T cells in concentrations in a range 1-5 µM. [11] while for dasatinib is 5-10 nM [21]. Results of Fei et al. [20] studies on nilotinib impact on Tregs revealed that nilotinib inhibited the proliferation and suppressive capacity of Tregs but in a dose-dependent manner. The production of cytokines secreted by Tregs and CD4+CD25- effector T cells was only inhibited at high concentrations of nilotinib exceeding the mean therapeutic serum concentrations of the drug in patients. Although, these results showed that nilotinib did not attenuate the function of Tregs at clinical relevant doses, this finding might be utilized after allogeneic stem cell transplantation.
Introduction of second generation kinase inhibitors to Ph+ hematological diseases has revolutionized therapy of CML and ALL. Although responses can be achieved in many patients using a second-generation TKI after the failure of imatinib or other therapy, the responses have still tended to be short-lived in some patients. Especially refractory patients, such as those harboring T315I mutation, have needed new agents for their therapy. Therefore there is fervent need for further development of new generation TKI.
Bosutinib
Bosutinib originally termed SKI-606, similarly to dasatinib is a dual Abl-Src kinase inhibitor. This Src kinase inhibitor was able to inhibit Fgr and Lyn TK even at picomolar concentrations. Further studies have revealed that it also targets Abl tyrosine kinase activity in low concentrations and could be more effective than imatinib but slightly weaker than dasatinib [63]. Bosutinib, in comparison to imatinib, could bind intermediate conformation of Abl [70] (tab. 1).
Table 1. Summarized features of tyrosine kinase inhibitors (TKI) [66,84]

Bosutinib was proved to be effective in several imatinib-resistant cell lines harboring different point mutants, the T315I mutation still remained undefeated. Bosutinib was effective as a second- and third-line treatment in patients who failed imatinib or dasatinib therapy.
Bosutinib is not yet approved by FDA for first-line therapy in chronic phase of CML. In phase I/II clinical trials, this compound yielded promising response rates, reduced toxicity and good tolerability in imatinib-resistant or -intolerant CML patients. 18-month evaluation of the BELA trial comparing bosutinib 500 mg/day with imatinib 400 mg/day in newly diagnosed patients presented at the European Hematology Association’s Annual Congress 2011 in London has been very promising. Faster and deepen responses were observed in patient treated with bosutinib than imatinib with fewer events of progression. The drug was well tolerated as long as initial gastrointestinal and liver side effects were managed well – with dose modifications or temporary interruptions [23]. In comparison to imatinib and dasatinib, bosutinib has only weak activity against PDGFR and KIT. This fact could explain safety since many side effects associated with imatinib such as edema, skin rash or inhibition of normal hemopoiesis have been related to the inhibition of PDGFR and/or KIT. Interestingly, bosutinib is active against a broader spectrum of kinases than originally was thought. Beside above mentioned ABL and SRC kinases this TKI targets TEC family kinases, which play a pivotal role in the regulation of the immune functions, especially BTK. Many of them, for example EPHB4, FER, PTK2 (FAK), PYK2, SYK and TNK2 are also often overexpressed in human cancers and are associated with poor prognosis. Also some kinases important for signal transduction, including MAPK signaling pathway are sensitive to bosutinib inhibition. Importantly, this compound is the first kinase inhibitor shown to target Ca2t/calmodulin-dependent protein (CAMK2G), which has been recently linked to myeloid leukemia cell proliferation. CAMK2G is involved in regulating signaling pathways controlling the proliferation of myeloid leukemia cells. Moreover, bosutinib is probably the first compound, among CAMK2G inhibitors, directly binding the adenosine triphosphate binding site of CAMK2G [71,78].
Since interactions between ABC multidrug transporters and several first-generation, small-molecule TKI are important in pharmacokinetics scientists have studied influence of MDR-ABC transporters on bosutinib. Hegedűs et al. [31] have found that neither ABCB1 nor ABCG2 interact with bosutinib at therapeutical concentration. These data have shown that multidrug resistance ATP-binding cassette (MDR-ABC) transporters cannot be responsible for bosutinib resistance and ABCB1 and ABCG2 could only slightly modify the cytotoxic effect of bosutinib.
Next generations of tyrosine kinase inhibitiors
Despite the great progress in the treatment of CML thanks to above described TKIs, scientists still have been searching for new, more effective and more potent agents, especially toward resistant mutants, such as T315I. The first novel potent inhibitor of BCR/ABL and aurora kinases (Aks) that shows clinical activity against T315I mutated form of BCR/ABL was MK-0457 [25]. Unfortunately this compound exerted serious cardiac toxicity [87]. Next novel TKI could be ponatinib (AP24534) and danusertib (formerly PHA-739358) [57]. Danusertib is a small molecule 3-aminopyrazole derivative that additionally inhibits some other cancer relevant TK such as wild type and mutated ABL, RET, TRK-A and FGFRs. Gontarewicz et al. [26] in their publication revealed potent antiproliferative effects of PHA-739358 in CD34+ cells derived from untreated and imatinib-resistant CML patients in chronic phase or blast crisis, including those harboring the T315I mutation. Recently, in phase I clinical trials danusertib has been tested in patient with solid tumors. The multicenter phase II study has recruited 12 relapsing CML patients. Objective clinical responses to danusertib have been observed in two CML patients with T315I mutations of BCR/ABL, with an acceptable tolerability and safety profile [60]. The aurora kinases family inhibitor ponatinib has demonstrated a potent activity against an wide array of BCR/ABL variants including T315I mutants and could also target other therapeutically relevant kinases such as FLT3, FGF, VEGF and PDGF, and c-KIT [28]. This agent is currently in a pivotal Phase II clinical trial as a treatment for CML patients or Ph+ ALL resistant to second generation TKIs. Ponatinib was very well tolerated until the determined maximum tolerated dose of 45 mg daily is not exceeded. This compound seems to be very promising as a second- or third-line treatment for patients with refractory CML and ALL, especially those harboring multidrug-resistant mutations of BCR/ABL.
Summarizing, as we have shown in this review, TKI have revolutionized CML therapies in a very short period of time. They have also a broader therapeutic array and could be used in PH+ ALL and GIST patients. TKI still represent one of the most interesting anticancer compounds giving a hope for a cure of patient. Better understanding of genetic alterations and oncogenic pathways in combination with knowledge about proprieties of TKI could lead to modifications of the structure of these compounds that could alter the mechanism of action improving the effectiveness of these agents.
REFERENCES
[1] Aichberger K.J., Mayerhofer M., Krauth M.T., Vales A., Kondo R., Derdak S., Pickl W.F., Selzer E., Deininger M., Druker B.J., Sillaber C., Esterbauer H., Valent P.: Low-level expression of proapoptotic Bcl-2-interacting mediator in leukemic cells in patients with chronic myeloid leukemia: role of BCR/ABL, characterization of underlying signaling pathways, and reexpression by novel pharmacologic compounds. Cancer Res., 2005; 65: 9436-9444
[PubMed] [Full Text HTML] [Full Text PDF]
[2] Al Achkar W., Wafa A., Mkrtchyan H., Moassass F., Liehr T.: A rare case of chronic myeloid leukemia with secondary chromosomal changes including partial trisomy 17q21 to 17qter and partial monosomy of 16p13.3. Mol. Cytogenet., 2010; 3: 6
[PubMed] [Full Text HTML] [Full Text PDF]
[3] Arora A., Scholar E.M.: Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther., 2005; 315: 971-979
[PubMed] [Full Text HTML] [Full Text PDF]
[4] Belli C., Alú M.F., Alfonso G., Bianchini M., Larripa I.: Novel variant Ph translocation t(9;22;11)(q34;q11.2;p15)inv(9)(p13q34) in chronic myeloid leukemia involving a one-step mechanism. Cytogenet. Genome Res., 2011; 132: 304-308
[PubMed]
[5] Belloc F., Moreau-Gaudry F., Uhalde M., Cazalis L., Jeanneteau M., Lacombe F., Praloran V., Mahon F.X.: Imatinib and nilotinib induce apoptosis of chronic myeloid leukemia cells through a Bim-dependant pathway modulated by cytokines. Cancer Biol. Ther., 2007; 6: 912-919
[PubMed] [Full Text PDF]
[6] Blake S.J., Bruce Lyons A., Fraser C.K., Hayball J.D., Hughes T.P.: Dasatinib suppresses in vitro natural killer cell cytotoxicity. Blood, 2008; 111: 4415-4416
[PubMed] [Full Text HTML] [Full Text PDF]
[7] Breccia M., Alimena G.: Resistance to imatinib in chronic myeloid leukemia and therapeutic approaches to circumvent the problem. Cardiovasc. Hematol. Disord. Drug Targets, 2009; 9: 21-28
[PubMed]
[8] Breccia M., Cannella L., Stefanizzi C., Carotti A., Santopietro M., Alimena G.: Efficacy of dasatinib in a chronic myeloid leukemia patient with disease molecular relapse and chronic GVHD after haploidentical BMT: an immunomodulatory effect? Bone Marrow Transplant., 2009; 44: 331-332
[PubMed]
[9] Buchdunger E., Cioffi C.L., Law N., Stover D., Ohno-Jones S., Druker B.J., Lydon N.B.: Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther., 2000; 295: 139-145
[PubMed] [Full Text HTML] [Full Text PDF]
[10] Carlson R.H.: CML: Dasatinib-related lymphocytosis associated with improved response in DASISION subanalysis. Oncology Times UK, 2011; 33: 31[abstract number 358]
[11] Chen J., Schmitt A., Giannopoulos K., Chen B., Rojewski M., Döhner H., Bunjes D., Schmitt M.: Imatinib impairs the proliferation and function of CD4+CD25+ regulatory T cells in a dose-dependent manner. Int. J. Oncol., 2007; 31: 1133-1139
[PubMed] [Full Text PDF]
[12] Chen Y., Peng C., Li D., Li S.: Molecular and cellular bases of chronic myeloid leukemia. Protein Cell, 2010; 1: 124-132
[PubMed]
[13] Deenik W., van der Holt B., Janssen J.J., Chu I.W., Valk P.J., Ossenkoppele G.J., van der Heiden I.P., Sonneveld P., van Schaik R.H., Cornelissen J.J.: Polymorphisms in the multidrug resistance gene MDR1 (ABCB1) predict for molecular resistance in patients with newly diagnosed chronic myeloid leukemia receiving high-dose imatinib. Blood, 2010; 116: 6144-6145
[PubMed]
[14] Deremer D.L., Katsanevas K., Ustun C.: Critical appraisal of nilotinib in frontline treatment of chronic myeloid leukemia. Cancer Manag. Res., 2011; 3: 65-78
[PubMed] [Full Text HTML] [Full Text PDF]
[15] Donato N.J., Wu J.Y., Stapley J., Gallick G., Lin H., Arlinghaus R., Talpaz M.: BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood, 2003; 101: 690-698
[PubMed] [Full Text HTML] [Full Text PDF]
[16] Druker B.J., Lydon N.B.: Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J. Clin. Invest., 2000; 105: 3-7
[PubMed] [Full Text HTML] [Full Text PDF]
[17] Duesberg P., Stindl R., Hehlmann R.: Origin of multidrug resistance in cells with and without multidrug resistance genes: chromosome reassortments catalyzed by aneuploidy. Proc. Natl. Acad. Sci. USA, 2001; 98: 11283-11288
[PubMed] [Full Text HTML] [Full Text PDF]
[18] Essafi A., Fernández de Mattos S., Hassen Y.A., Soeiro I., Mufti G.J., Thomas N.S., Medema R.H., Lam E.W.: Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl-expressing cells. Oncogene, 2005; 24: 2317-2329
[PubMed]
[19] Fausel C.: Targeted chronic myeloid leukemia therapy: seeking a cure. J. Manag. Care Pharm., 2007; 13 (Suppl. A): 8-12
[PubMed] [Full Text PDF]
[20] Fei F., Yu Y., Schmitt A., Rojewski M.T., Chen B., Greiner J., Götz M., Bunjes D., Schmitt M.: Effects of nilotinib on regulatory T cells: the dose matters. Mol. Cancer, 2010; 9: 22
[PubMed] [Full Text HTML] [Full Text PDF]
[21] Fei F., Yu Y., Schmitt A., Rojewski M.T., Chen B., Greiner J., Götz M., Guillaume P., Döhner H., Bunjes D., Schmitt M.: Dasatinib exerts an immunosuppressive effect on CD8+ T cells specific for viral and leukemia antigens. Exp. Hematol., 2008; 36: 1297-1308
[PubMed]
[22] Foa R., Vitale A., Guarini A., De Propris A.S., Elia R., Cimino G., Luppi M., Castagnola C., Sica S., Nieddu R., Piersantelli M., Ferrara F., Nobile F., Fanin R., Fabbiano F., Vignetti M., Fazi P., Soverini S., Mandelli F., Martinelli G., Meloni G., Baccarani M.: Dasatinib monotherapy effective and feasible as first-line treatment of adult Philadelphia-chromosome positive acute lymphoblastic leukemia: Final results of the GIMEMA LAL1205 study. Blood, 2008; 112: 119 [abstract number 305]
[23] Gambacorti-Passerini C., Cortes J.E., Kim D., Kantarjian H., Khattry N., Lipton J.H., Powell C., Harris P., Countouriotis A.M., Brummendorf T.H.: Bosutinib (BOS) versus imatinib (IM) in patients (pts) with chronic phase chronic myeloid leukemia (CP CML) in the BELA trial: 18-month follow-up. J. Clin. Oncol., 2011; 29 [abstract number 6509]
[24] Giannoudis A., Davies A., Lucas C.M., Harris R.J., Pirmohamed M., Clark R.E.: Effective dasatinib uptake may occur without human organic cation transporter 1 (hOCT1): implications for the treatment of imatinib-resistant chronic myeloid leukemia. Blood, 2008; 112: 3348-3354
[PubMed] [Full Text HTML]
[25] Giles F.J., Cortes J., Jones D., Bergstrom D., Kantarjian H., Freedman S.J.: MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood, 2007; 109: 500-502
[PubMed] [Full Text HTML] [Full Text PDF]
[26] Gontarewicz A., Balabanov S., Keller G., Colombo R., Graziano A., Pesenti E., Benten D., Bokemeyer C., Fiedler W., Moll J., Brümmendorf T.H.: Simultaneous targeting of Aurora kinases and Bcr-Abl kinase by the small molecule inhibitor PHA-739358 is effective against imatinib-resistant BCR-ABL mutations including T315I. Blood, 2008; 111: 4355-4364
[PubMed] [Full Text HTML] [Full Text PDF]
[27] Gorre M.E., Mohammed M., Ellwood K., Hsu N., Paquette R., Rao P.N., Sawyers C.L.: Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science, 2001; 293: 876-880
[PubMed]
[28] Gozgit J.M., Wong M.J., Wardwell S., Tyner J.W., Loriaux M.M., Mohemmad Q.K., Narasimhan N.I., Shakespeare W.C., Wang F., Druker B.J., Clackson T., Rivera V.M.: Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol. Cancer Ther., 2011; 10: 1028-1035
[PubMed]
[29] Griffin J.D., Todd R.F. 3rd, Ritz J., Nadler L.M., Canellos G.P., Rosenthal D., Gallivan M., Beveridge R.P., Weinstein H., Karp D., Schlossman S.F.: Differentiation patterns in the blastic phase of chronic myeloid leukemia. Blood, 1983; 61: 85-91
[PubMed] [Full Text PDF]
[30] Guerrouahen B.S., Futami M., Vaklavas C., Kanerva J., Whichard Z.L., Nwawka K., Blanchard E.G., Lee F.Y., Robinson L.J., Arceci R., Kornblau S.M., Wieder E., Cayre Y.E., Corey S.J.: Dasatinib inhibits the growth of molecularly heterogeneous myeloid leukemias. Clin. Cancer Res., 2010; 16: 1149-1158
[PubMed] [Full Text HTML] [Full Text PDF]
[31] Hegedus C., Ozvegy-Laczka C., Apáti A., Magócsi M., Német K., Orfi L., Kéri G., Katona M., Takáts Z., Váradi A., Szakács G., Sarkadi B.: Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol., 2009; 158: 1153-1164
[PubMed] [Full Text HTML] [Full Text PDF]
[32] Heinrich M.C., Griffith D.J., Druker B.J., Wait C.L., Ott K.A., Zigler A.J.: Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood, 2000; 96: 925-932
[PubMed] [Full Text HTML] [Full Text PDF]
[33] Hiwase D.K., Saunders V., Hewett D., Frede A., Zrim S., Dang P., Eadie L., To L.B., Melo J., Kumar S., Hughes T.P., White D.L.: Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clin. Cancer Res., 2008; 14: 3881-3888
[PubMed] [Full Text HTML] [Full Text PDF]
[34] Hochhaus A., Kreil S., Corbin A.S., La Rosée P., Müller M.C., Lahaye T., Hanfstein B., Schoch C., Cross N.C., Berger U., Gschaidmeier H., Druker B.J., Hehlmann R.: Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia, 2002; 16: 2190-2196
[PubMed] [Full Text HTML] [Full Text PDF]
[35] Hochhaus A., La Rosée P.: Imatinib therapy in chronic myelogenous leukemia: strategies to avoid and overcome resistance. Leukemia, 2004; 18: 1321-1331
[PubMed]
[36] Hoelbl A., Schuster C., Kovacic B., Zhu B., Wickre M., Hoelzl M.A., Fajmann S., Grebien F., Warsch W., Stengl G., Hennighausen L., Poli V., Beug H., Moriggl R., Sexl V.: Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol. Med., 2010; 2: 98-110
[PubMed] [Full Text HTML] [Full Text PDF]
[37] Hrycyna C.A.: Molecular genetic analysis and biochemical characterization of mammalian P-glycoproteins involved in multidrug resistance. Semin. Cell Dev. Biol., 2001; 12: 247-256
[PubMed]
[38] Hrycyna C.A., Ramachandra M., Germann U.A., Cheng P.W., Pastan I., Gottesman M.M.: Both ATP sites of human P-glycoprotein are essential but not symmetric. Biochemistry, 1999; 38: 13887-13899
[PubMed]
[39] Hunter T.: The role of tyrosine phosphorylation in cell growth and disease. Harvey Lect., 1998-1999; 94: 81-119
[PubMed]
[40] Illmer T., Schaich M., Platzbecker U., Freiberg-Richter J., Oelschlägel U., von Bonin M., Pursche S., Bergemann T., Ehninger G., Schleyer E.: P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia, 2004; 18: 401-408
[PubMed]
[41] Johansson B., Fioretos T., Mitelman F.: Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol., 2002; 107: 76-94
[PubMed]
[42] Kamath A.V., Wang J., Lee F.Y., Marathe P.H.: Preclinical pharmacokinetics and in vitro metabolism of dasatinib (BMS-354825): a potent oral multi-targeted kinase inhibitor against SRC and BCR-ABL. Cancer Chemother. Pharmacol., 2008; 61: 365-376
[PubMed]
[43] Kantarjian H., Shah N.P., Hochhaus A., Cortes J., Shah S., Ayala M., Moiraghi B., Shen Z., Mayer J., Pasquini R., Nakamae H., Huguet F., Boqué C., Chuah C., Bleickardt E., Bradley-Garelik M.B., Zhu C., Szatrowski T., Shapiro D., Baccarani M.: Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med., 2010; 362: 2260-2270
[PubMed]
[44] Kantarjian H.M., Cortes J.E., O’Brien S., Luthra R., Giles F., Verstovsek S., Faderl S., Thomas D., Garcia-Manero G., Rios M.B., Shan J., Jones D., Talpaz M.: Long-term survival benefit and improved complete cytogenetic and molecular response rates with imatinib mesylate in Philadelphia chromosome-positive chronic-phase chronic myeloid leukemia after failure of interferon-α. Blood, 2004; 104: 1979-1988
[PubMed] [Full Text HTML] [Full Text PDF]
[45] Konig H., Copland M., Chu S., Jove R., Holyoake T.L., Bhatia R.: Effects of dasatinib on SRC kinase activity and downstream intracellular signaling in primitive chronic myelogenous leukemia hematopoietic cells. Cancer Res., 2008; 68: 9624-9633
[PubMed]
[46] Kreutzman A., Juvonen V., Kairisto V., Ekblom M., Stenke L., Seggewiss R., Porkka K., Mustjoki S.: Mono/oligoclonal T and NK cells are common in chronic myeloid leukemia patients at diagnosis and expand during dasatinib therapy. Blood, 2010; 116: 772-782
[PubMed] [Full Text HTML] [Full Text PDF]
[47] Kubota T.: Gastrointestinal stromal tumor (GIST) and imatinib. Int. J. Clin. Oncol., 2006; 11: 184-189
[PubMed]
[48] Larson R.A., Druker B.J., Guilhot F., O’Brien S.G., Riviere G.J., Krahnke T., Gathmann I., Wang Y.: Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood, 2008; 111: 4022-4028
[PubMed] [Full Text HTML] [Full Text PDF]
[49] Mahon F.X., Hayette S., Lagarde V., Belloc F., Turcq B., Nicolini F., Belanger C., Manley P.W., Leroy C., Etienne G., Roche S., Pasquet J.M.: Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res., 2008; 68: 9809-9816
[PubMed] [Full Text HTML] [Full Text PDF]
[50] Manley P.W., Cowan-Jacob S.W., Mestan J.: Advances in the structural biology, design and clinical development of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid leukaemia. Biochim. Biophys. Acta, 2005; 1754: 3-13
[PubMed]
[51] Manley P.W., Drueckes P., Fendrich G., Furet P., Liebetanz J., Martiny-Baron G., Mestan J., Trappe J., Wartmann M., Fabbro D.: Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim. Biophys. Acta, 2010; 1804: 445-453
[PubMed]
[52] Mauro M.J., Druker B.J.: STI571: targeting BCR-ABL as therapy for CML. Oncologist, 2001; 6: 233-238
[PubMed] [Full Text HTML] [Full Text PDF]
[53] Moriggl R., Sexl V., Kenner L., Duntsch C., Stangl K., Gingras S., Hoffmeyer A., Bauer A., Piekorz R., Wang D., Bunting K.D., Wagner E.F., Sonneck K., Valent P., Ihle J.N., Beug H.: Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell, 2005; 7: 87-99
[PubMed] [Full Text HTML] [Full Text PDF]
[54] Mustjoki S., Ekblom M., Arstila T.P., Dybedal I., Epling-Burnette P.K., Guilhot F., Hjorth-Hansen H., Höglund M., Kovanen P., Laurinolli T., Liesveld J., Paquette R., Pinilla-Ibarz J., Rauhala A., Shah N., Simonsson B., Sinisalo M., Steegmann J.L., Stenke L., Porkka K..: Clonal expansion of T/NK-cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia, 2009; 23: 1398-1405
[PubMed]
[55] Nam S., Williams A., Vultur A., List A., Bhalla K., Smith D., Lee F.Y., Jove R.: Dasatinib (BMS-354825) inhibits Stat5 signaling associated with apoptosis in chronic myelogenous leukemia cells. Mol. Cancer Ther., 2007; 6: 1400-1405
[PubMed] [Full Text HTML] [Full Text PDF]
[56] O’Hare T., Shakespeare W.C., Zhu X., Eide C.A., Rivera V.M., Wang F., Adrian L.T., Zhou T., Huang W.S., Xu Q., Metcalf C.A. 3rd, Tyner J.W., Loriaux M.M., Corbin A.S., Wardwell S., Ning Y., Keats J.A., Wang Y., Sundaramoorthi R., Thomas M., Zhou D., Snodgrass J., Commodore L., Sawyer T.K., Dalgarno D.C., Deininger M.W., Druker B.J., Clackson T.: AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell, 2009; 16: 401-412
[PubMed] [Full Text HTML] [Full Text PDF]
[57] O’Hare T., Walters D.K., Stoffregen E.P., Jia T., Manley P.W., Mestan J., Cowan-Jacob S.W., Lee F.Y., Heinrich M.C., Deininger M.W., Druker B.J.: In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res., 2005; 65: 4500-4505
[PubMed] [Full Text HTML] [Full Text PDF]
[58] Olivieri A., Manzione L.: Dasatinib: a new step in molecular target therapy. Ann. Oncol., 2007; 18 (Suppl. 6): vi42-vi46
[PubMed] [Full Text PDF]
[59] Ottmann O.G., Druker B.J., Sawyers C.L., Goldman J.M., Reiffers J., Silver R.T., Tura S., Fischer T., Deininger M.W., Schiffer C.A., Baccarani M., Gratwohl A., Hochhaus A., Hoelzer D., Fernandes-Reese S., Gathmann I., Capdeville R., O’Brien S.G.: A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood, 2002; 100: 1965-1971
[PubMed] [Full Text HTML] [Full Text PDF]
[60] Paquette R., Shah N., Sawyers C., Martinelli G., Nicoll J.: Pha-739358: a pan-aurora kinase inhibitor. Hematology Meeting Reports, 2008; 2: 92-93
[61] Pene-Dumitrescu T., Smithgall T.E.: Expression of a Src family kinase in chronic myelogenous leukemia cells induces resistance to imatinib in a kinase-dependent manner. J. Biol. Chem., 2010; 285: 21446-21457
[PubMed] [Full Text HTML] [Full Text PDF]
[62] Pui C.H., Jeha S.: New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat. Rev. Drug Discov., 2007; 6: 149-165
[PubMed]
[63] IPuttini M., Coluccia A.M., Boschelli F., Cleris L., Marchesi E., Donella-Deana A., Ahmed S., Redaelli S., Piazza R., Magistroni V., Andreoni F., Scapozza L., Formelli F., Gambacorti-Passerini C.: In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res., 2006; 66: 11314-11322
[PubMed] [Full Text HTML] [Full Text PDF]
[64] Quintas-Cardama A., Kantarjian H., Talpaz M., O’Brien S., Garcia-Manero G., Verstovsek S., Rios M.B., Hayes K., Glassman A., Bekele B.N., Zhou X., Cortes J.: Imatinib mesylate therapy may overcome the poor prognostic significance of deletions of derivative chromosome 9 in patients with chronic myelogenous leukemia. Blood, 2005; 105: 2281-2286
[PubMed] [Full Text HTML] [Full Text PDF]
[65] Radich J.P.: Philadelphia chromosome-positive acute lymphocytic leukemia. Hematol. Oncol. Clin. North Am., 2001; 15: 21-36
[PubMed]
[66] Rajaraman S., Davis W.S., Mahakali-Zama A., Evans H.K., Russell L.B., Bedell M.A.: An allelic series of mutations in the Kit ligand gene of mice. II. Effects of ethylnitrosourea-induced Kitl point mutations on survival and peripheral blood cells of Kitl(Steel) mice. Genetics, 2002; 162: 341-353
[PubMed] [Full Text HTML] [Full Text PDF]
[67] Ravandi F.: Dasatinib, an immunomodulator? Blood, 2010; 116: 673-674
[PubMed]
[68] Ravandi F., O’Brien S., Thomas D., Faderl S., Jones D., Garris R., Dara S., Jorgensen J., Kebriaei P., Champlin R., Borthakur G., Burger J., Ferrajoli A., Garcia-Manero G., Wierda W., Cortes J., Kantarjian H.: First report of phase 2 study of dasatinib with hyper-CVAD for the frontline treatment of patients with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia. Blood, 2010; 116: 2070-2077
[PubMed] [Full Text HTML] [Full Text PDF]
[69] Ray A., Cowan-Jacob S.W., Manley P.W., Mestan J., Griffin J.D.: Identification of BCR-ABL point mutations conferring resistance to the Abl kinase inhibitor AMN107 (nilotinib) by a random mutagenesis study. Blood, 2007; 109: 5011-5015
[PubMed] [Full Text HTML] [Full Text PDF]
[70] Redaelli S., Piazza R., Rostagno R., Magistroni V., Perini P., Marega M., Gambacorti-Passerini C., Boschelli F.: Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J. Clin. Oncol., 2009; 27: 469-471
[PubMed]
[71] Remsing Rix L.L., Rix U., Colinge J., Hantschel O., Bennett K.L., Stranzl T., Müller A., Baumgartner C., Valent P., Augustin M., Till J.H., Superti-Furga G.: Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia, 2009; 23: 477-485
[PubMed]
[72] Rudduck-Sivaswaren C., Tien S.L., Lim P., Lim E., Lie D.K., Tan P.H., Lee A.S.: Evidence for deletion of 9q as a two-step process in chronic myeloid leukemia. Clin. Genet., 2005; 68: 461-465
[PubMed]
[73] Sacha T.: Molekularne mechanizmy oporności na imatinib. Acta Haematol. Pol., 2003; 34: 263-275
[Abstract] [Full Text PDF]
[74] Sawyers C.L., Hochhaus A., Feldman E., Goldman J.M., Miller C.B., Ottmann O.G., Schiffer C.A., Talpaz M., Guilhot F., Deininger M.W., Fischer T., O’Brien S.G., Stone R.M., Gambacorti-Passerini C.B., Russell N.H., Reiffers J.J., Shea T.C., Chapuis B., Coutre S., Tura S., Morra E., Larson R.A., Saven A., Peschel C., Gratwohl A., Mandelli F., Ben-Am M., Gathmann I., Capdeville R., Paquette R.L., Druker B.J.: Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood, 2002; 99: 3530-3539
[PubMed] [Full Text HTML] [Full Text PDF]
[75] Schade A.E., Schieven G.L., Townsend R., Jankowska A.M., Susulic V., Zhang R., Szpurka H., Maciejewski J.P.: Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood, 2008; 111: 1366-1377
[PubMed] [Full Text HTML] [Full Text PDF]
[76] Shah N.P., Nicoll J.M., Nagar B., Gorre M.E., Paquette R.L., Kuriyan J., Sawyers C.L.: Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell, 2002; 2: 117-125
[PubMed] [Full Text HTML] [Full Text PDF]
[77] Shah N.P., Tran C., Lee F.Y., Chen P., Norris D., Sawyers C.L.: Overriding imatinib resistance with a novel ABL kinase inhibitor. Science, 2004; 305: 399-401
[PubMed]
[78] Si J., Collins S.J.: Activated Ca2+/calmodulin-dependent protein kinase IIγ is a critical regulator of myeloid leukemia cell proliferation. Cancer Res., 2008; 68: 3733-3742
[PubMed] [Full Text HTML] [Full Text PDF]
[79] Soverini S., Colarossi S., Gnani A., Rosti G., Castagnetti F., Poerio A., Iacobucci I., Amabile M., Abruzzese E., Orlandi E., Radaelli F., Ciccone F., Tiribelli M., di Lorenzo R., Caracciolo C., Izzo B., Pane F., Saglio G., Baccarani M., Martinelli G.: Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin. Cancer Res., 2006; 12: 7374-7379
[PubMed]
[80] Steinberg M.: Dasatinib: a tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Clin. Ther., 2007; 29: 2289-2308
[PubMed]
[81] Talpaz M., Shah N.P., Kantarjian H., Donato N., Nicoll J., Paquette R., Cortes J., O’Brien S., Nicaise C., Bleickardt E., Blackwood-Chirchir M.A., Iyer V., Chen T.T., Huang F., Decillis A.P., Sawyers C.L.: Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med., 2006; 354: 2531-2541
[PubMed] [Full Text HTML] [Full Text PDF]
[82] Talpaz M., Silver R.T., Druker B.J., Goldman J.M., Gambacorti-Passerini C., Guilhot F., Schiffer C.A., Fischer T., Deininger M.W., Lennard A.L., Hochhaus A., Ottmann O.G., Gratwohl A., Baccarani M., Stone R., Tura S., Mahon F.X., Fernandes-Reese S., Gathmann I., Capdeville R., Kantarjian H.M., Sawyers C.L.: Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood, 2002; 99: 1928-1937
[PubMed] [Full Text HTML] [Full Text PDF]
[83] Tokarski J.S., Newitt J.A., Chang C.Y., Cheng J.D., Wittekind M., Kiefer S.E., Kish K., Lee F.Y., Borzillerri R., Lombardo L.J., Xie D., Zhang Y., Klei H.E.: The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res., 2006; 66: 5790-5797
[PubMed] [Full Text HTML] [Full Text PDF]
[84] Tsao A.S., Kantarjian H., Cortes J., O’Brien S., Talpaz M.: Imatinib mesylate causes hypopigmentation in the skin. Cancer, 2003; 98: 2483-2487
[PubMed] [Full Text HTML] [Full Text PDF]
[85] Wadleigh M., DeAngelo D.J., Griffin J.D., Stone R.M.: After chronic myelogenous leukemia: tyrosine kinase inhibitors in other hematologic malignancies. Blood, 2005; 105: 22-30
[PubMed] [Full Text HTML] [Full Text PDF]
[86] Wassmann B., Pfeifer H., Scheuring U.J., Binckebanck A., Gökbuget N., Atta J., Brück P., Rieder H., Schoch C., Leimer L., Schwerdtfeger R., Ehninger G., Lipp T., Perz J., Stelljes M., Gschaidmeier H., Hoelzer D., Ottmann O.G.: Early prediction of response in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ALL) treated with imatinib. Blood, 2004; 103: 1495-1498
[PubMed] [Full Text HTML] [Full Text PDF]
[87] Wei G., Rafiyath S., Liu D.: First-line treatment for chronic myeloid leukemia: dasatinib, nilotinib, or imatinib. J. Hematol. Oncol., 2010; 3: 47
[PubMed] [Full Text HTML] [Full Text PDF]
[88] Weisberg E., Manley P.W., Breitenstein W., Brüggen J., Cowan-Jacob S.W., Ray A., Huntly B., Fabbro D., Fendrich G., Hall-Meyers E., Kung A.L., Mestan J., Daley G.Q., Callahan L., Catley L., Cavazza C., Azam M., Neuberg D., Wright R.D., Gilliland D.G., Griffin J.D.: Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell, 2005; 7: 129-141
[PubMed] [Full Text HTML] [Full Text PDF]
[89] White D.L., Saunders V.A., Dang P., Engler J., Venables A., Zrim S., Zannettino A., Lynch K., Manley P.W., Hughes T.: Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood, 2007; 110: 4064-4072
[PubMed] [Full Text HTML] [Full Text PDF]
[90] Wilson M.B., Schreiner S.J., Choi H.J., Kamens J., Smithgall T.E.: Selective pyrrolo-pyrimidine inhibitors reveal a necessary role for Src family kinases in Bcr-Abl signal transduction and oncogenesis. Oncogene, 2002; 21: 8075-8088
[PubMed] [Full Text HTML] [Full Text PDF]
[91] Wisniewski D., Lambek C.L., Liu C., Strife A., Veach D.R., Nagar B., Young M.A., Schindler T., Bornmann W.G., Bertino J.R., Kuriyan J., Clarkson B.: Characterization of potent inhibitors of the Bcr-Abl and the c-kit receptor tyrosine kinases. Cancer Res., 2002; 62: 4244-4255
[PubMed] [Full Text HTML] [Full Text PDF]
[92] Wolff N.C., Veach D.R., Tong W.P., Bornmann W.G., Clarkson B., Ilaria R.L.Jr: PD166326, a novel tyrosine kinase inhibitor, has greater antileukemic activity than imatinib mesylate in a murine model of chronic myeloid leukemia. Blood, 2005; 105: 3995-4003
[PubMed] [Full Text HTML] [Full Text PDF]
[93] Wu J., Meng F., Lu H., Kong L., Bornmann W., Peng Z., Talpaz M., Donato N.J.: Lyn regulates BCR-ABL and Gab2 tyrosine phosphorylation and c-Cbl protein stability in imatinib-resistant chronic myelogenous leukemia cells. Blood, 2008; 111: 3821-3829
[PubMed] [Full Text HTML] [Full Text PDF]
[94] Yu H., Jove R.: The STATs of cancer-new molecular targets come of age. Nat. Rev. Cancer, 2004; 4: 97-105
[PubMed]
The authors have no potential conflicts of interest to declare.