
Postępy w kierunku molekularnej klasyfikacji nowotworów u dzieci*
Joanna Szczepanek 1 , Jan Styczyński 2 , Olga Haus 3 , Andrzej Tretyn 4 , Mariusz Wysocki 2Streszczenie
Nowotwory najczęściej są klasyfikowane zgodnie z charakterystyką histologiczną, lokalizacją lub występowaniem określonych markerów molekularnych. Rozwój nowych technologii spowodował potrzebę opracowania skutecznej i uniwersalnej metody analizy, umożliwiającej zarówno ustalanie typów nowotworów jak i przyporządkowywanie nowych przypadków do odpowiednich grup ryzyka. Technika mikromacierzy stanowi uzupełnienie konwencjonalnych metod analitycznych. Pozwala na znaczne skrócenie czasu od pobrania materiału do analiz do otrzymania precyzyjnej charakterystyki komórek nowotworowych. Molekularna klasyfikacja w oparciu o technikę mikromacierzy odzwierciedla biologiczną i kliniczną różnorodność chorób nowotworowych. Istnieje możliwość określenia profilu ekspresji pozwalającego na dokładną diagnozę, ocenę stopnia zaawansowana, stratyfikację pacjentów do grup ryzyka oraz dobór właściwego postępowania terapeutycznego dla poszczególnych nowotworów. Strategie takie zastosowano już w badaniach niektórych nowotworów dziecięcych, takich jak białaczki, chłoniaki i mięsaki. W pracy zaprezentowano możliwości zastosowania mikromacierzy w tworzenie nowych klasyfikacji najczęstszych chorób nowotworowych wieku dziecięcego.
Słowa kluczowe:profil ekspresji • mikromacierze • klasyfikacja • nowotwory u dzieci • transkryptomika
Summary
Neoplasms are usually classified according to their histology, location, or the occurrence of specific molecular markers. The development of new technologies has led to the necessity of applying more efficient and universal methods of analysis, enabling a better diagnosis of the type of cancer and the determination of risk groups. Microarray technology constitutes the completion of conventional analytical methods. It shortens the time for a complete and precise characterization of cancer cells. Molecular classification based on microarray technology reflects the biological and clinical variety of neoplastic diseases. Gene expression profiles enable precise diagnosis, scoring and grading, and risk factor stratification. This technique might thus lead to targeted therapy in each type of cancer. This type of strategy has already been used in several types of pediatric cancer, such as leukemia, lymphoma, and sarcoma. In this review the possibilities of a microarray- based classification of pediatric cancers are presented.
Key words:expression profile • microarrays • classification • cancers in children • transcriptomic
WPROWADZENIE
W ciągu ostatnich kilkudziesięciu lat znacznie usprawniono diagnostykę i terapię przeciw nowotworom złośliwym u dzieci. Jest to możliwe dzięki zastosowaniu nowych metod diagnozowania obrazowego, histologicznego i immunologicznego oraz wprowadzeniu metod genetyki molekularnej. Tradycyjna klasyfikacja nowotworów posługuje się głównie rozpoznaniem histopatologicznym [31]. Diagnostyka nowotworów hematologicznych coraz częściej opiera się, poza badaniem morfologicznym, na nowoczesnych technikach, takich jak cytometria przepływowa, immunohistochemia oraz cytogenetyka. Znacznie trudniejsza jest analiza guzów litych [19].
Stosowany system klasyfikacji chorób nowotworowych jest jednak wciąż niezadowalający ze względu na to, iż do większości wznów dochodzi w grupach standardowego ryzyka. Precyzyjny system klasyfikacji powinien umożliwiać w czasie rozpoznania przyporządkowanie chorych do odpowiedniej grupy ryzyka oraz dobór najodpowiedniejszej strategii leczenia. Wynika stąd potrzeba rozwoju i ulepszania stratyfikacji chorób nowotworowych, a tym samym umożliwienie zindywidualizowanej i zoptymalizowanej terapii. Skuteczny system klasyfikacji powinien umożliwiać wyodrębnienie z dużej, a zarazem różnorodnej grupy mniejszej, homogennej podgrupy o podobnej biologii i prognozie przebiegu choroby [18].
Duże nadzieje pokłada się w nowoczesnych metodach systeomiki, pozwalających na złożone, kompleksowe analizy na różnych poziomach ekspresji informacji w komórce. Głównymi narzędziami analizy zarówno genomu (poziom DNA), transkryptomu (poziom RNA) oraz proteomu (poziom białek) są mikromacierze [21,104]. Techniki te pozwalają na opracowywanie „molekularnych portretów” komórek, co ma ogromne znaczenie zwłaszcza w przypadku heterogennych grup nowotworów. Molekularna klasyfikacja stwarza możliwość odróżniania histologicznie podobnych nowotworów, usprawniając tym samym diagnozę oraz stratyfikację pacjentów.
PROFILE EKSPRESJI GENÓW
Szybki postęp w genetyce molekularnej znacznie zrewolucjonizował poznawanie biologii chorób nowotworowych. Nowe techniki, oparte na profilowaniu ekspresji genów oraz analizach zmian w obrębie całego genomu, budzą nadzieje na opracowanie nowoczesnego, czułego i swoistego systemu klasyfikacji [53]. Technika mikromacierzy łączy w sobie zalety takich technik jak: cytogenetyka, hybrydyzacja in situ oraz RT-PCR.
Klasyfikacją nowotworów w oparciu o profile ekspresji genów jako jedni z pierwszych zajmowali się Golub i wsp. [31]. Badacze ci wprowadzili do użytku dwa terminy: „odkrywania klas” (class discovery) – identyfikacji nieznanych podtypów nowotworów oraz „przewidywania klas” (class prediction) – przyporządkowywania nowotworów do zdefiniowanych grup [31]. Ważnym aspektem klasyfikacji nowotworów jest ponadto „porównywanie klas” (class comparison) rozumiane jako zestawianie cech profilu ekspresji w obrębie dobrze zdefiniowanych już grup [18].
Najpopularniejszą techniką profilowania jest monitorowanie poziomu transkrypcyjnego RNA (mRNA). Dzięki macierzom ekspresyjnym możliwe jest analizowanie w jednym doświadczeniu danych dotyczących poziomu ekspresji wielu tysięcy genów, a nawet całej znanej sekwencji genomu. W badaniach tkanek nowotworowych najczęściej wykorzystywane są macierze cDNA (komplementarnego DNA) oraz macierze oligonukleotydowe GeneChip™ (Affymetrix) [41,84]. Dzięki tej technice możliwa jest identyfikacja licznych grup genów. Często do dalszych analiz (klasyfikacji, prognozowania) wystarczające są dane dotyczące niektórych z wytypowanych genów i możliwe jest badanie ich innymi, tańszymi i mniej skomplikowanymi, metodami biologii molekularnej (np. QRT-PCR).
Główne zastosowanie metody mikromacierzy w badaniach nad chorobami nowotworowymi obejmuje:
• molekularną charakterystykę komórek patologicznych,
• identyfikację nowych klas nowotworów, niewyodrębnionych za pomocą aktualnie stosowanych metod diagnostycznych,
• opracowanie molekularnego systemu klasyfikacji na podstawie profilu ekspresji,
• porównywanie podtypów nowotworów,
• udoskonalenie stratyfikacji pacjentów do grup ryzyka za pomocą nowych zestawów genów,
• na podstawie identyfikacji genów związanych z lekoopornością opracowanie programów terapii celowanej oraz nowych celów molekularnych dla leków [71,90,91,100].
SYSTEMY KLASYFIKACJI STOSOWANE W ONKOLOGII DZIECIĘCEJ
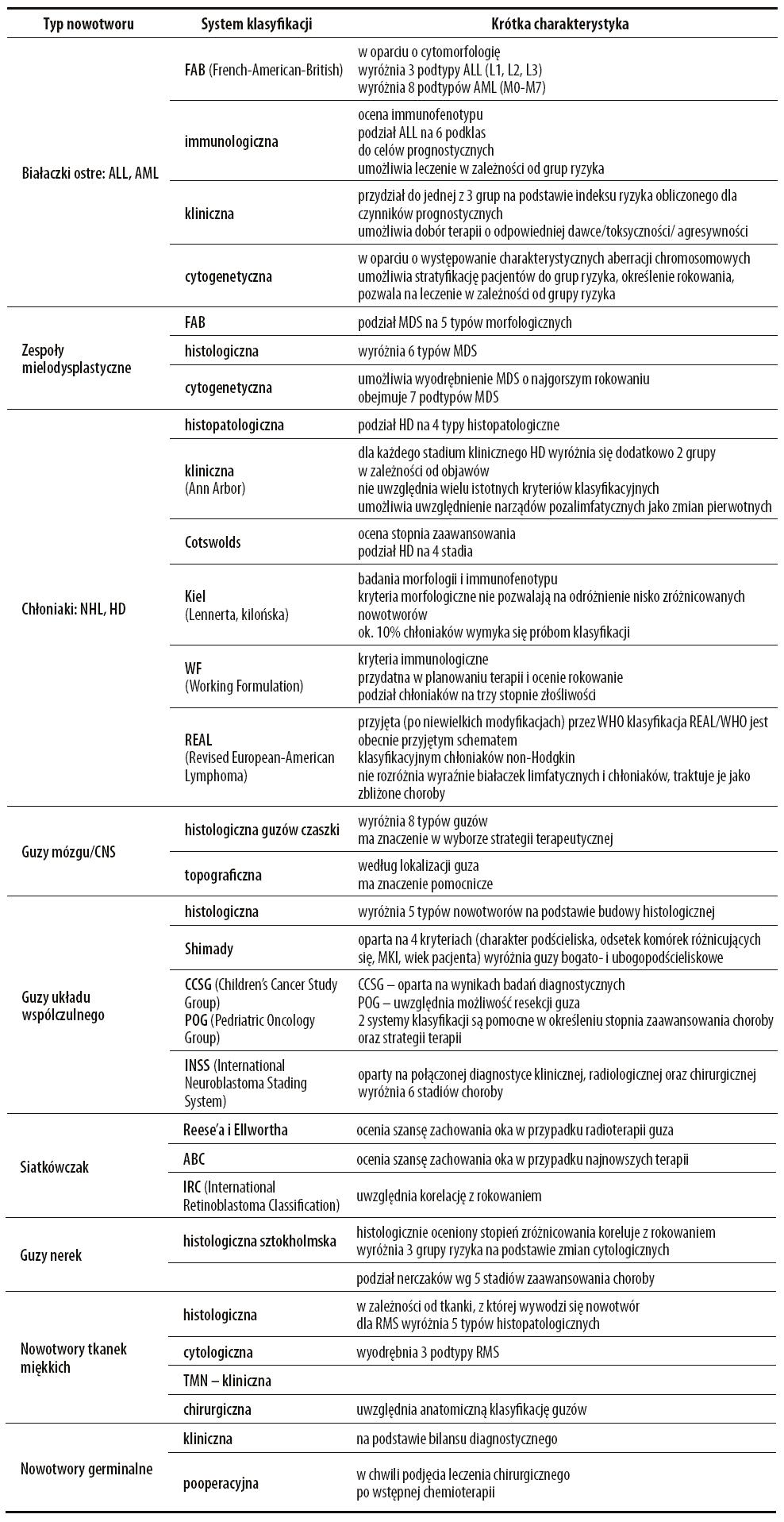
W onkologii dziecięcej wciąż funkcjonuje wiele systemów klasyfikacyjnych (tabela 1). W związku z tym, iż każda korzysta z innych kryteriów podziału, służą innym celom klinicznym. Poza klasyfikacjami odnoszącymi się do poszczególnych nowotworów stworzono również uniwersalne systemy dla wszystkich typów nowotworów jak ICCC (International Classification of Childhood Cancers) oraz klasyfikacja WHO (The World Health Organization Classification of Tumors) (tabela 3).
Tabela 1. Systemy klasyfikacji stosowane w onkologii dziecięcej
Tradycyjna klasyfikacja nowotworów opiera się głównie na rozpoznaniu histopatologicznym, a taki system jest zdecydowanie niezadowalający. Precyzyjny system klasyfikacji powinien umożliwiać w chwili diagnozy przyporządkowanie do odpowiedniej grupy ryzyka oraz dobór najodpowiedniejszej strategii terapeutycznej. Alternatywę dla tych wszystkich systemów stanowić może, zyskujące coraz większe uznanie klasyfikowanie na podstawie profilowania ekspresji z wykorzystaniem mikromacierzy (tabela 2). Technika ta oferuje, w pojedynczym doświadczeniu, szeroki zakres przeanalizowanych genów oraz możliwości korelacji otrzymanych wyników z istotnym cechami klinicznymi w onkologii. Z powodzeniem jest stosowana w analizach porównawczych nowotworów u dzieci, takich jak białaczki czy mięsaki.
Tabela 2. Porównanie klasyfikacji tradycyjnej i molekularnej

Jej podstawowymi zaletami są wysokie współczynniki czułości, swoistości i dokładności – często wyższe w porównaniu z tradycyjnymi metodami diagnostycznymi. Dzięki profilowaniu ekspresji genów możliwe jest precyzyjne odróżnianie nowotworów o podobnej charakterystyce histologicznej, takich jak np. guzy drobnookrągłoniebieskokomórkowe (SRBC). Profilowanie ekspresji genów stwarza możliwość dokładnej charakterystyki typów nowotworów określonych innymi metodami w innych systemach klasyfikacji (tabela 3). Zastosowanie tej techniki ma także kolejna zaletę, jaką jest skrócenie czasu od chwili przyjęcia pacjenta na oddział do postawienia prawidłowej diagnozy i doboru odpowiedniej terapii, co ma podstawowe znaczenie w onkologii.
Tabela 3. Porównanie głównych uniwersalnych systemów klasyfikacji nowotworów występujących u dzieci

NOWOTWORY WYSTĘPUJĄCE U DZIECI
Nowotwory wieku dziecięcego występują rzadziej niż u dorosłych. Stanowią prawie 1% ogółu diagnozowanych chorób nowotworowych [81]. Jednak jako druga przyczyna zgonów w tej grupie wiekowej (po wypadkach i zatruciach) są poważnym problemem współczesnej medycyny [39]. W Polsce co roku na nowotwory zapada około 1200 dzieci. Rodzaj i częstość występowania nowotworów u dzieci są inne niż u dorosłych, co jest konsekwencją odmiennego przebiegu klinicznego, biologii i uwarunkowań genetycznych. Szansę na trwałe wyleczenie ma ponad 70% pacjentów pediatrycznych. Prawie 50% nowotworów u dzieci stanowią nowotwory hematologiczne (białaczki i chłoniaki), druga połowa to guzy lite (ryc. 1) [20]. Zgodnie z klasyfikacją ICD-O-3 (International Classification of Diseases for Oncology) oraz ICCC-3 (International Classification of Childhood Cancers) wśród nowotworów wieku dziecięcego wyróżnia się 12 głównych grup diagnostycznych oraz 47 podgrup [85]. Terminem „guzy lite” u dzieci obejmuje się przede wszystkim: guzy ośrodkowego układu nerwowego, nerwiaki (neuroblastoma), mięsaki (sarcoma) tkanek miękkich, nerczaki (nephroblastoma – guz Wilmsa), guzy kości (osteosarcoma), siatkówczaki (retinoblastoma), wątrobiaki (hepatoblastoma) oraz guzy zarodkowe (ryc. 1) [81].

Ryc. 1. Częstotliwość występowania nowotworów dziecięcych w Polsce (na podstawie [48], zmodyfikowane)
PRÓBY ZASTOSOWANIA PROFILOWANIA EKSPRESJI W KLASYFIKACJI NOWOTWORÓW DZIECIĘCYCH
Nowotwory hematologiczne
Określenie nowotwory hematologiczne obejmuje białaczki, chłoniaki, szpiczaka mnogiego oraz zespoły mielodysplastyczne [68]. Obecny system klasyfikacji jest oparty na połączeniu danych z analizy morfologii, cytochemii, kariotypu, immunofenotypu oraz molekularnych cech komórek [35,41,45,59]. W 2001 r. WHO opublikowała ujednolicony system klasyfikacji nowotworów mieloidalnych i limfoidalnych, oparty na licznych wynikach analiz wielu grup badawczych w ciągu kilku ostatnich lat [8]. Technika mikromacierzy jest szeroko stosowana w licznych analizach tej grupy chorób nowotworowych wieku dziecięcego. Dzięki niej dokonano znacznego postępu przede wszystkim w diagnostyce i klasyfikacji, ale również w prognozowaniu i terapii [7,31,75,79,86,103]. W niedalekiej przyszłości klasyfikacja nowotworów hematologicznych za pomocą profili ekspresji może się stać cennym narzędziem diagnostycznym [6,35,63,86]. Przeszkodę stanowią wciąż zbyt duże zestawy identyfikowanych genów, stosowanych jako molekularne klasyfikatory [63].
Ostre białaczki
Ostre białaczki (acute leukemia – AL), najczęstszy typ nowotworów u dzieci, charakteryzują się występowaniem zaburzeń genetycznych, które definiują poszczególne podklasy AL. Stanowią niejednorodną grupę chorób nowotworowych. Przede wszystkim wyróżnia się dwie grupy w zależności od linii komórek, z których się wywodzą, tj. ostre białaczki limfoblastyczne (acute lymphoblastic leukemia – ALL) oraz mieloblastyczne (szpikowe, acute myeloblastic leukemia – AML). Możliwe jest przeprowadzenie podziału ostrych białaczek na podstawie klasyfikacji: br• morfologicznej: opracowana przez zespół francuskoamerykańsko- brytyjski (FAB), w latach 70 ub.w, stopniowo modyfikowana, dzieli AML na osiem podtypów (M0–M7), a ALL na trzy podtypy (L1–L3),
• cytochemicznej: stanowi uzupełnienie danych morfologicznych o liczne reakcje, np. analizy aktywności peroksydazy (POX) czy swoistych esteraz,
• immunologicznej: oparta na analizie antygenów różnicowania komórkowego (cluster of differentation – CD) z wykorzystaniem przeciwciał monoklonalnych, odgrywa rolę w odróżnianiu ALL od AML, rozpoznawaniu białaczki megakariocytowej oraz diagnozowaniu podtypów ALL z prekursorów linii B- lub T-komórkowej oraz dojrzałych limfocytów B i T,
• cytogenetycznej: analiza liczbowych i strukturalnych zmian chromosomowych, z użyciem cytogenetyki klasycznej oraz molekularnej, np. fluorescencyjnej hybrydyzacji in situ (fluorescence in situ hybridization – FISH),
• molekularnej: badanie zmian w strukturze DNA, na poziomie RNA oraz produktów genów. Techniki stosowane w tego rodzaju badaniach (np. PCR, RT-PCR, RQ-PCR) umożliwiają precyzyjne rozpoznawanie i klasyfikowanie nowotworów (tabela 4) [12,26].
Tabela 4. Klasyfikacja ostrych białaczek limfoblastycznych

Najsilniejsze czynniki prognostyczne opierają się na parametrach cytogenetycznych i molekularnych (typu: hiperdiploidia, rearanżacje TEL-AML1, BCR-ABL, MLL) oraz odpowiedzi na terapię (steroidooporność iv vivo, czas uzyskania remisji, obecność minimalnej choroby resztkowej). Obecnie dąży się do opracowanie schematu klasyfikacji w oparciu o zmiany cytogenetyczne i molekularne [35].
W licznych analizach wykazano powiązanie profili ekspresji genów z występowaniem określonych cech immunofenotypowych czy zmian genetycznych [7,42,44,77,103]. Określono również wzorce ekspresji genów, a tym samym grupy genów różnicujących umożliwiających precyzyjną stratyfikację pacjentów [5,45,77,103]. W badaniach wciąż wykorzystywane są różne typy chipów DNA oraz stosowane są odmienne algorytmy analizy danych, dlatego też często występują rozbieżności w wynikach analiz różnych zespołów badawczych (tabela 5). Niemniej jednak analizy prowadzone przez różne zespoły badawcze są stopniowo coraz bardziej zbieżne (np. grupy genów różnicujących określonych dla 132 przypadków wszystkich podtypów genetycznych ALL [77] oraz 40 linii komórkowych [4] pokryły się prawie w 80%) [5,63,76,77].
Tabela 5. Zestawienie liczby genów różnicujących, umożliwiających klasyfikację głównych podtypów B-liniowej ALL

W diagnostyce i terapii ostrych białaczek podstawowe znaczenie ma rozpoznanie linii komórek, z których rozwinęła się choroba. Stosowane metody pozwalają na dość precyzyjne odróżnienie pacjentów z ALL od pacjentów z AML. Niewątpliwym utrudnieniem jest jednak konieczność przeprowadzenia w tym celu kilku testów. Golub i wsp. [31], jako pierwsi zwrócili uwagę, że alternatywę dla wielu analiz stanowić może monitorowanie setek genów za pomocą mikromacierzy oligonukleotydowych. Pomiędzy analizowanymi dwoma typami ostrych białaczek istnieją różnice w ekspresji około 1100 genów, przy czym 50 najbardziej reprezentatywnych genów wystarczyło do klasyfikowania nowych przypadków ALL i AML. Wśród omawianej grupy genów znalazły się zarówno dobrze znane markery, jak i takie, których dalsze analizy mogą potwierdzić istotny udział w procesie zezłośliwienie blastów i pozwolą na uznanie ich jako markerów. W obrębie AML odnotowano większą różnorodność profili ekspresji w porównaniu z ALL [63].
Ostra białaczka limfoblastyczna
Jak wykazano w licznych badaniach za pomocą techniki chipów DNA można z dużą dokładnością klasyfikować ostre białaczki jako ALL i AML [7,31], a także wyróżniać B-liniową i T-liniową ALL [6,37,77,98,103]. Golub i wsp. [31] wykazali, iż molekularna klasyfikacja pacjentów w oparciu o profile ekspresji genów umożliwia odkrywanie i przewidywanie typów ALL lub AML bez wiedzy a priori o nich. Zasugerowali też, iż nowe klasy i podtypy ostrych białaczek mogą mieć biologiczne oraz kliniczne znaczenie [31]. Willenbrock i wsp. [98] wykazali, iż na podstawie wzorca ekspresji 29 genów możliwe jest oznaczenie immunofenotypu komórek nowotworowych ze 100% dokładnością, nawet w przypadkach, gdy liczba limfoblastów jest na poziomie 10%. W wielu analizach potwierdzono, że na podstawie profilu ekspresji genów w limfoblastach można precyzyjnie klasyfikować poszczególne cytogenetyczne podtypy ALL: z występowaniem hiperdiploidii (HD) >50 chromosomów, t(12;21)(TEL-AML1), 11q23/MLL, t(1;19)(E2A-PBX1), t(9;22)(BCR-ABL), a nawet identyfikować nowe grupy (tabela 5) [77,103]. BCR-ABL ALL stanowi najbardziej heterogenną podgrupę B-komórkowej ALL [5,29,63]. Za pomocą profilu ekspresji genów precyzyjnie klasyfikowano przypadki ALL zgodnie z cechami histologicznymi i cytogenetycznymi. W wielu przypadkach jako molekularne klasyfikatory identyfikowano grupy genów o dotychczas nieznanej roli w powstawaniu i przebiegu ALL [29,36,77]. Wykazano, iż taka klasyfikacja koreluje z rokowaniem w ALL, zarówno B- jak i T-komórkowej [41,77,103]. Badania te potwierdzono na niezależnych grupach pacjentów. W wielu przypadkach identyfikowano dla poszczególnych podtypów setki genów, których zmiany w ekspresji świadczą o znaczących różnicach w globalnym profilu ekspresji limfoblastów. Do celów wyłącznie klasyfikacji oraz oceny ryzyka, często wystarczająca jest informacja o ekspresji od kilku do kilkudziesięciu genów o najwyższej wartości predykcyjnej oraz odpowiednio czułe algorytmy analizy danych. Analiza ekspresji zaledwie jednego genu jest wystarczająca do określenia fenotypu T-ALL (CD3D) lub B-ALL rearanżacją E2A-PBX (PBX1) ze 100% dokładnością. Stratyfikacja pacjentów do pozostałych podtypów wymaga analizy koekspresji 7–20 genów [103]. Moos i wsp. [57] opracowali zestawy, zawierające po 20 genów dyskryminujących, pozwalających na odróżnienie pacjentów z ALL od AML, z B- od T-liniowej ALL, ALL z grupy standardowego i wysokiego ryzyka, a także B-ALL z rearanżacją TEL-AML1 od przypadków bez obecności translokacji.
Na podstawie analiz profili ekspresji genów wywnioskowano, iż przypadki ostrych białaczek, którym towarzyszy rearanżacja MLL, powinny być klasyfikowane jako odmienny od ALL i AML typ białaczki [7]. Dotychczasowa klasyfikacja pacjentów z tego typu aberracją była utrudniona ze względu na to, iż towarzyszy jej koekspresja markerów charakterystycznych zarówno dla linii limfoidalnej jak i mieloidalnej [7,19]. Profil ekspresji genów w przypadku białaczek z rearanżacją MLL jest na tyle charakterystyczny, iż pozwala na stratyfikację pacjentów z 97% dokładnością [7]. Z powodzeniem określono również profile ekspresji dla podtypów T-ALL, jak LYL1, HOX11, TAL1, a także nowego podtypu z rearanżacją HOX11L2 [28]. Również w obrębie B-ALL zidentyfikowano nową podgrupę mającą wyraźnie odmienny profil ekspresji, a niewykazującej żadnej aberracji cytogenetycznej [77,103].
Informacja o ekspresji wyselekcjonowanych 20 genów może służyć precyzyjnej identyfikacji grupy pacjentów z wysokim ryzykiem wznowy, zwłaszcza w obrębie immunofenotypu T-ALL oraz HD>50. Za pomocą innego zestawu 20 genów można wyodrębnić grupę pacjentów, u których istnieje wysokie prawdopodobieństwo wystąpienie indukowanej terapią AML [103] lub nowotworów mózgu (zestaw 33 genów) [27]. Gottardo i wsp. [32] zauważyli, iż na podstawie wzorca ekspresji 3 genów (biorących udział w apoptozie i proliferacji – CFLAR, NOTCH2, BTG3) można określić rokowanie w dziecięcej T-ALL.
Willenbrock i wsp. [98] analizowali komórki białaczkowe pod kątem możliwości stratyfikacji pacjentów do odpowiednich grup ryzyka na podstawie wzorca ekspresji genów. W zależności od liczby typowanych genów różnicujących (2-45) oraz stosowanej metody analizy otrzymali różne poziomy dokładności i swoistości. Poziom ten jednak wyniósł prawie 70%, co i tak jest wyższą wartością w porównaniu z konwencjonalną klasyfikacją grup ryzyka, na podstawie wieku pacjenta, początkowej leukocytozy czy immunofenotypu. Nieco dokładniejsze wyniki w przypadku stratyfikacji pacjentów do grup wysokiego lub niskiego ryzyka otrzymali Teuffel i wsp. [89]. Za jedne z najważniejszych cech dyskryminujących grupę wysokiego ryzyka uznali zmiany w ekspresji 6 genów dla białek rybosomalnych, cykliny H oraz genu proapoptotycznego LRDD/Pidd. Nie udało się jednak opracować profilu ekspresji dla grupy pośredniego ryzyka, wówczas gdy analizie poddano bardzo zróżnicowaną grupę pacjentów. Teuffel i wsp. [89] podkreślają, że obecnie profil ekspresji genetycznej jest niewystarczający do precyzyjnej i niezależnej od innych czynników stratyfikacji pacjentów. Z tego też względu opinie na temat czy sam profil ekspresji jest wystarczający dla takiej stratyfikacji i czy może być alternatywą lub uzupełnieniem dla obecnych kryteriów stratyfikacji ALL są wciąż podzielone.
Jak wynika z kilku przeprowadzonych analiz, we wzorcu ekspresji genów jest odzwierciedlona również prognoza odpowiedzi na leczenie indukcyjne i konsolidacyjne [17]. Dotychczas ukazało się kilka prac omawiających możliwości zastosowania macierzy ekspresyjnych w optymalizacji i doborze odpowiedniej strategii leczenia. Na podstawie profilu ekspresji genów możliwe jest stwierdzenie w czasie diagnozy, u których pacjentów wystąpi wrażliwość lub oporność na dany lek, a nawet kombinację kilku leków [17,36,37,54].
Podkreśla się przede wszystkim wysoką czułość i swoistość stratyfikacji pacjentów na podstawie profilu molekularnego [77]. Wielokrotnie wykazano, iż taki system klasyfikacji jest dokładniejszy oraz dostarcza więcej użytecznych informacji w porównaniu z klasycznymi technikami laboratoryjnymi. W badaniach Yeoha i wsp. [103] oraz Ross i wsp. [77] osiągnięto 96% dokładność stratyfikacji w obrębie 6 prognostycznie istotnych typów ALL u dzieci. A zatem precyzja klasyfikacji za pomocą samych mikromacierzy jest porównywalna z dokładnością aktualnej klasyfikacji z wykorzystaniem kilku metod jednocześnie.
Ostra białaczka mieloblastyczna
Nowotwory należące do tej grupy wywodzą się z wczesnych etapów różnicowania układu granulocytowo-monocytowego oraz erytroblastycznego. Najpowszechniej stosowany w klasyfikacji AML system FAB, oparty na cytomorfologii, obejmuje podtypy M0–M7 (tabela 6) [101]. W ciągu kilku ostatnich lat WHO uzupełniła ten schemat o dane cytogenetyczne i molekularne. W klasyfikacji WHO wyróżnia się 4 główne typy AML (tabela 6). Główną wadą tego sytemu jest brak bezpośredniej relacji między występującymi nieprawidłowościami kariotypu oraz wzrastająca liczbą nowych markerów cytogenetycznych a prognozą [16].
Tabela 6. Klasyfikacja ostrych białaczek mieloblastycznych wg FAB i WHO

Obecny podział grup ryzyka w AML odbywa się na podstawie danych cytogenetycznych oraz wczesnej odpowiedzi na terapię [40]. Z punktu widzenia terapii w ostrej białaczce mieloblastycznej szczególne znaczenie ma klasyfikacja pacjentów zaliczanych do grupy standardowego ryzyka, stanowiącej prawie połowę diagnozowanych przypadków. Obecnie trudnością jest to, iż w tej grupie występuje duża różnorodność zmian cytogenetycznych, a ponadto w licznych przypadkach stwierdza się prawidłowy kariotyp [33,58]. Wielu badaczy podjęło się próby utworzenia nowej klasyfikacji AML w oparciu o profile ekspresji [15,34,92]. Profil ekspresji genów przede wszystkim umożliwia przyporządkowanie poszczególnych przypadków do odpowiednich podtypów AML w klasyfikacji FAB [34,35]. W najwcześniejszych analizach badacze wykazali, iż na podstawie profilu ekspresji genów możliwe jest odróżnianie AML z obecnością trisomii chromosomu 8 od przypadków AML o kariotypie niezmienionym [94]. Późniejsze analizy doprowadziły do opracowania wzorców ekspresji dla 3 najważniejszych podtypów AML, tj.: t(15;17)(PML-RARa), t(8;21)(AML1-ETO) oraz inv(16)(CBFβ-MYH11) [79]. Badaczom udało się również stwierdzić, iż na podstawie wzorca ekspresji określonego zestawu genów możliwe jest identyfikowanie z dużą dokładnością grup pacjentów z określonym rokowaniem (przypadki z inv(16), t(8;21), t(15;17), mutacjami w CEBPA) [14,24,34,44,76,79,93]. Jak wskazują analizy przeprowadzone przez Ross i wsp. [76], na podstawie wyselekcjonowanych grup genów można przewidywać podtypy AML z 93% dokładnością. Takie molekularne klasyfikatory opracowano dla pięciu podtypów ostrej białaczki mieloblastycznej: PML-RARa, AML1-ETO, CBFβ-MYH11, z rearanżacją MLL oraz podtypu M7 w klasyfikacji FAB [76]. Valk i wsp. [93] po przeanalizowaniu zróżnicowanej pod względem cytogenetycznym grupy 285 pacjentów, stwierdzili występowanie 16 odmiennych wzorców ekspresji genów w obrębie AML. Z przeprowadzonych badań wynika, iż największa różnorodność profili ekspresji dotyczy AML z prawidłowym kariotypem, rearanżacją 11q23, trisomią chromosomu 8 lub monosomią chromosomu 7 [14,93,94].
Jak opisują Bullinger i wsp. [14] takie metody badań genetycznych w połączeniu z odpowiednimi algorytmami analizy danych mogą w konsekwencji doprowadzić do wyodrębnienia nowych podgrup AML u dorosłych. Dwie nowe zidentyfikowane w ten sposób grupy cechowały się brakiem zmian w kariotypie, ale u pacjentów stwierdzono różną długość przeżycia. Profilowanie ekspresji może również pomóc w wyodrębnianiu pacjentów, u których stwierdza się często obecność tych samych markerów prognostycznych do grup standardowego i wysokiego ryzyka [16]. Bullinger i wsp. [14] zidentyfikowali 133 geny, których zmiany w profilu ekspresji odpowiadały grupom ryzyka u dorosłych, wyznaczonym metodami cytogenetycznymi. Z kolei Yagi i wsp. [101] jako klasyfikator, służący przyporządkowaniu pacjentów pediatrycznych do grup niskiego lub wysokiego ryzyka, użyli zestawu 35 genów. Badacze ci podkreślają, iż większość z zidentyfikowanych genów nie była dotychczas uznawana za markery prognostyczne, nie była również wiązana z klasyfikacją FAB czy znanymi rearanżacjami chromosomowymi. Profile ekspresji opracowane przez omawiane zespoły badawcze dla dwóch grup wiekowych były w dużej mierze zbieżne [14,101]. Jak wynika z licznych analiz silna korelacja wzorca genetycznego z prognozą i rokowaniem powinna stanowić podstawę reklasyfikacji [14,51,93,101].
Chociaż przeprowadzone analizy już stanowią znaczny postęp w rozumieniu złożonej patobiologii AML, do stworzenia pełnego obrazu komórek nowotworowych należy rozważyć wprowadzenie nowych formatów biochipów, takich jak: mikromacierze CGH (porównawczej hybrydyzacji genomowej do mikromacierzy), mikromacierze dla wysp CpG, mikromacierze z zestawami przeciwciał oraz inne techniki analiz całego genomu [16].
Zespoły mielodysplastyczne
Zespoły mielodysplastyczne (myelodysplastic syndrome – MDS) stanowią grupę chorób charakteryzującą się przede wszystkim cytopenią, zaburzeniami hematopoezy oraz występowaniem cech dysplazji w szpiku kostnym. Wynik leczenia uzależniony jest przede wszystkim od zaawansowania choroby, stąd potrzeba molekularnej charakterystyki odzwierciedlającej stadium rozwoju MDS. Brakuje danych dotyczących znaczenia badań na skalę całego genomu w MDS u dzieci. W jedynej przeprowadzonej analizie Roela i wsp. [72] użyli techniki mikromacierzy w badaniu pediatrycznych przypadków MDS. Jak zaobserwowali, prawidłowe komórki szpiku kostnego oraz komórki patologiczne w przypadku MDS oraz AML związanej z MDS mają wyraźnie odmienne profile ekspresji. Dla każdej z grup zidentyfikowali zestawy kilkudziesięciu genów dobrze każdą z nich charakteryzujące i umożliwiające porównywanie pomiędzy nimi. Na przykład w profilu ekspresji mogącym odróżnić MDS od AML-MDS charakterystyczne było wyciszanie ekspresji genów biorących udział w endocytozie oraz sekrecji białek.
Choroba Hodgkina i chłoniaki nieziarnicze
Nieziarnicze chłoniaki złośliwe (non-Hodgkin lymphoma – NHL) stanowią grupę nowotworów wywodzących się z różnych stadiów różnicowania limfocytów B i T oraz komórek naturalnej cytotoksyczności (NK). Z kolei choroba Hodgkina (Hodgkin disease) jest nowotworem tkanki limfoidalnej, którego cechą charakterystyczną jest obecność patologicznych komórek Reed-Sternberga oraz prekursorowych komórek Hodgkina. Klasyfikacja nowotworów limfoproliferacyjnych stwarza największe trudności we współczesnej onkologii [18]. Diagnostyka nieziarniczych chłoniaków złośliwych oraz ziarnicy złośliwej oparta jest na zmodyfikowanej klasyfikacji histopatologicznej REAL/WHO (Revised European-American Classification of Lymphoid Neoplasms – REAL) (tabele 7, 8) [18,26].
Tabela 7. Główne histopatologiczne kategorie chłoniaków non-Hodgkin u dzieci (wg [70])

Tabela 8. Klasyfikacje chłoniaka Hodgkina (wg [47])

Podobnie jak w przypadku białaczek diagnostyka wymaga przeprowadzenia kompleksowych analiz histopatologicznych i genetycznych.
Morfologia chłoniaków jest uzależniona od stopnia zróżnicowania, występowania zmian genetycznych oraz licznych procesów fizjologicznych zachodzących w układzie chłonnym. Istnieje zatem potrzeba udoskonalenia klasyfikacji histologicznej oraz potrzeba zastąpienia lub uzupełnienia metod diagnostyki cytogenetycznej w celu zoptymalizowania standardów diagnostycznych [47]. Wiele grup badawczych podejmowało również próby molekularnej klasyfikacji chłoniaków na podstawie profilowania ekspresji. W analizie komórek chłoniaków wykorzystywane są techniki:
• immunofenotypowania z zastosowaniem mikromacierzy z przeciwciałami, służące przede wszystkim identyfikacji antygenów powierzchniowych leukocytów [10,11],
• analizy jednoczesnej profilu białkowego metodą macierzy proteinowych [30],
• określania profilu ekspresji genów z zastosowaniem mikromacierzy ekspresyjnych [2,3,74,83].
Morfologia chłoniaków zależy od stopnia zróżnicowania oraz stanu czynnościowego komórek. W związku ze znaczną liczbą zjawisk zachodzących fizjologicznie w układzie chłonnym, morfologia i cechy czynnościowe chłoniaków należą do najbardziej zróżnicowanych wśród nowotworów. Stąd też konieczność rozbudowy i doprecyzowania klasyfikacji histologicznej, w oparciu o nowe metody diagnostyczne.
Charakterystyka choroby Hodgkina na skalę całego genomu wskazuje na możliwość identyfikacji trzech molekularnych podtypów w jej obrębie. Określone na podstawie profilowania ekspresji genów grupy są związane histologią oraz odpowiedzią na terapię i przeżyciem. Devilard i wsp. [25] określili odrębne zestawy genów różnicujących dla jednej podklasy o złym oraz dwóch o dobrym rokowaniu. Analizowane przypadki HD o lepszym przebiegu klinicznym wyróżniały się przede wszystkim wysokim poziomem ekspresji genów proapoptotycznych oraz uczestniczących w przekazywaniu sygnałów w komórce. Znaczny spadek liczby transkryptów genów zaangażowanych w aktywację fibroblastów, angiogenezę, proliferację komórek oraz genów supresorowych obserwowano u pacjentów, u których mimo standardowej terapii dochodziło do dalszego postępu choroby i śmierci [25]. Jak podkreślają autorzy cytowanej pracy, molekularna taksonomia choroby Hodgkina powiązana zarówno z efektywnością terapii jak i przebiegiem klinicznym, sugeruje możliwość usprawnienia bieżącej klasyfikacji.
Na podstawie informacji o genach ulegających aktywacji i wyciszaniu możliwe jest również klasyfikowanie chłoniaka Burkitta oraz chłoniaka rozlanego z dużych komórek B (diffuse large B-cell lymphoma – DLBCL) [22]. Dla 30% przypadków analizowanych przez Dave’a i wsp. [22] dane były rozbieżne z klasyfikacją histologiczną. Opracowane profile ekspresji genów umożliwiały odróżnianie na poziomie molekularnym chłoniaka Burkitta od każdego z podtypów DLBCL.
Nowotwory układu nerwowego
Guzy mózgu
Guzy mózgu u dzieci stanowią heterogenną grupę chorób (tabela 9, tabela 10), których diagnoza oraz przewidywanie rokowania na podstawie cech morfologicznych są utrudnione [69,78]. W dotychczasowych badaniach nowotworów rozwijających się w obrębie OUN opracowano profile ekspresji charakterystyczne dla różnych typów guzów, wielu podklas histologicznych, poszczególnych stadiów przerzutowania oraz pozwalające na ocenę rokowania [46].
Tabela 9. Skrócona wersja klasyfikacji histopatologicznej pierwotnych nowotworów OUN według Światowej Organizacji Zdrowia (WHO) z 1993 r.

Tabela 10. Klasyfikacja histologiczna nerczaka zarodkowego wg grup ryzyka, stosowana w protokole SIOP 2001 (wg [49])

Jednym z najczęstszych nowotworów mózgu u dzieci jest wyściółczak wewnątrzczaszkowy (intracranial ependymomas). Pod względem histologicznym tego typu guzy są trudno odróżnialne od innych nowotworów OUN. Klinicznie wykazują znaczną heterogenność. Korshunov i wsp. [46], na podstawie opracowanych profili ekspresji genów, byli w stanie klasyfikować poszczególne przypadki wyściółczaków zgodnie z ich lokalizacją, stopniem zaawansowania oraz wiekiem pacjenta. Dzięki analizie profilu ekspresji wyściółczaków z różnych części mózgu, Taylor i wsp. [88] udowodnili hipotezę, iż ten typ nowotworu wywodzi się z różnych komórek prekursorowych. Poszczególne wzorce ekspresji genów kojarzono z anatomiczną lokalizacją tkanki guza, nie stwierdzono jednak związku z innymi parametrami klinicznymi oraz z rozpoznaniem histologicznym. W grupie genów różnicujących znaczące były zmiany w poziomie transkryptów genów uczestniczących w regulacji proliferacji i różnicowania komórek prekursorowych danej części mózgu. Wyraźnie odmienne wzorce ekspresji mają wyściółczaki: obustronne (posterior fossa), nadnamiotowe (supratentorial) i kręgowe (spinal) [88].
Innym często diagnozowanym nowotworem mózgu u dzieci jest rdzeniak zarodkowy (medulloblastoma). Opracowany przez Pomeroya i wsp. [69] system klasyfikacji w oparciu o technikę mikromacierzy umożliwia molekularne odróżnienie tej grupy nowotworów od niedojrzałego guza neuroektodermalnego (primitive neuroectodermal tumor – PNET), a także atypowego guza teratoido/rabdoidalnego (atypical teratoid/rhabdoid tumour – AT/RT) oraz złośliwych glejaków (gliomas). Zgodnie z oczekiwaniami najbardziej odmienny profil ekspresji miały glejaki, których komórki mają pochodzenie nieneuronalne. Dlatego wzorzec ekspresji tej grupy nowotworów cechowała ekspresja genów charakterystycznych dla astrocytów i oligodendrocytów (PEA15, SOX2, PMP2, Olig-2, TrkB, S100, GFAP), genów związanych z metabolizmem (2,6-bifosfataza fruktozy, dehydrogenaza glutaminianu) oraz genów biorących udział w różnicowaniu komórek (ID2, GDF1, TYK2). W profilu ekspresji genów w komórkach rdzeniaka zarodkowego dominowała ekspresja genów czynników transkrypcyjnych, w tym ZIC i NSCL1, charakteryzujących komórki ziarniste móżdżku (cerebellar granule cells – CGCs). Jak sugerują Pomeroy i wsp. [69] powyższe dane wskazują, iż rdzeniak zarodkowy wywodzi się właśnie ze wspomnianej linii komórek lub w komórkach nowotworowych aktywacji ulega program transkrypcyjny swoisty dla CGCs. Badacze ci opracowali również profile ekspresji umożliwiające precyzyjną klasyfikację dwóch podklas w obrębie tego nowotworu. Doprowadzili także do wyselekcjonowania markerów różnicujących AT/RT od guzów rabdoidalnych. Wśród genów, których ekspresja była różna w przypadku tych dwu nowotworów znalazły się: szkieletowa β-tropomiozyna, NFAT3, lekki łańcuch miozyny.
Pomeroy i wsp. [69] potwierdzili również, iż na podstawie profilu ekspresji genów w komórkach nowotworowych cewiaka nerwowego, można określić rokowanie już podczas diagnozy. Jest to niewątpliwie znaczne usprawnienie niewystarczającego obecnie prognozowania w oparciu o stadium rozwoju nowotworu, ocenione na podstawie rozmiaru guza i obecności przerzutów. Na podstawie analiz profilu genetycznego zidentyfikowano geny korelujące z lepszym rokowaniem. W grupie znalazły się także takie geny jak: b-NAP, NSCL1, TRKC, geny kodujące białka kanałów sodowych, hydroksylazę PLOD, kolagen typu V αI, elastyna. Niekorzystne rokowanie stwierdzono u pacjentów, w komórkach których wzorzec ekspresji charakteryzowała obniżona ekspresja genów związanych z różnicowaniem komórek móżdżku. Wzorzec ekspresji w źle rokujących rdzeniakach zarodkowych zdominowany jest przez geny związane z proliferacją komórek, z opornością wielolekową oraz z metabolizmem (MYBL2, enolaza I, LDH, MHG1(Y), oksydaza cytochromu C), a także geny kodujące białka rybosomalne.
Nowotwory układu współczulnego – neuroblastoma
Wykonano również analizy profilu ekspresji dla nowotworowych neuroblastów [64,65,80]. W badaniach przeprowadzonych przez Krasnoselsky’ego i wsp. [50] sprawdzono możliwość wykorzystania profilu molekularnego w ocenie stopnia zaawansowania omawianego typu nowotworu. Stadium 4, bez amplifikacji genu MYCN, charakteryzowało się spadkiem poziomu transkryptów genów regulujących cykl komórkowy. Nadekspresja wspomnianej grupy genów występowała w stadium 3. Pomiędzy stadium 1 i 3 (bez amplifikacji MYCN) występowały różnice w poziomie ekspresji 309 genów. W neuroblastach w najwyższym, czwartym stopniu zaawansowania, z obecnością albo brakiem amplifikacji MYCN, istniały różnice w liczbie transkryptów 691 genów. Krasnoselsky i wsp. [50] stwierdzili również istnienie unikatowego wzorca ekspresji towarzyszącego występowaniu amplifikacji genu MYCN niezależnie od stadium zaawansowania neuroblastoma.
Klasyfikacja do grup ryzyka w obrębie neuroblastoma odbywa się zgodnie ze schematem Children’s Oncology Group Neuroblastoma Risk Group Assignment Schema UICC 2005 [97]. Jest ona niezadowalająca, zwłaszcza w obrębie grupy wysokiego ryzyka. Wei i wsp. [97], używając profilu ekspresji genów do przewidywania prognozy osiągnęli 88% dokładność. Wyodrębnili oni także zestaw 19 genów (w tym dwóch markerów prognostycznych – MYCN i CD44), za pomocą których można prognozować przebieg choroby i ustalać rokowanie u 98% pacjentów. Analiza transkryptomu neuroblastów umożliwia wyróżnienie dwóch podgrup pacjentów z różną długością przeżycia w grupie wysokiego ryzyka. Z badań Oberthuera i wsp. [64] wynika, iż klasyfikatorem dla różnych stopni ryzyka może być zestaw 144 genów. Ich analiza ekspresji pozwala na klasyfikowanie pacjentów do trzech grup ryzyka [58]. Warnat i wsp. [96] potwierdzili występowanie dwóch odmiennych biologicznie grup neuroblastoma. Określanie rokowania dla tego nowotwory na podstawie wzorca ekspresji zostało przeprowadzone z 77% dokładnością. Badacze ci uznali, iż analiza profilu ekspresji 77 genów różnicujących jest ważnym molekularnym wskaźnikiem agresywności guza [96].
Guzy nerek (nerczak zarodkowy, nerczak płodowy, nephroblastoma)
Najczęstszym złośliwym nowotworem nerek występującym u dzieci jest guz Wilmsa (nerczak zarodkowy) [99,106,107]. Stanowi on prawie 90% wszystkich guzów nerek oraz 7– 10% wszystkich nowotworów litych u dzieci [49]. W większości przypadków rokowania dla pacjentów są dobre. Na podstawie histologii wyróżnia się dwa główne podtypy guza Wilmsa: (1) pomyślnie rokujący oraz (2) z występowaniem anaplazji (tabela 10) [87]. Williams i wsp. [99] zastosowali technikę mikromacierzy cDNA do wyznaczenia grup pacjentów, u których istnieje wysokie prawdopodobieństwo wznowy. Pomiędzy dwoma analizowanymi grupami przypadków istniały różnice w profilu ekspresji 138 genów, przy czym przewidywanie wyników terapii jest możliwe na podstawie danych o ekspresji znacznie mniejszej liczby genów. Wyniki tych badań sugerują, że na ryzyko nawrotu choroby mają wpływ produkty takich genów jak: CCNI1, KRAS2, PGLYRP, BIRC6, dla których odnotowano wzrost liczby transkryptów. Złe rokowanie dla dzieci z tego typu nowotworem nerek wiąże się również ze spadkiem aktywności NOTCH1, FAF1, TNFRSF1A, GRIM19, IL10RB oraz BMP4. Chociaż udało się wyodrębnić pacjentów zgodnie z rokowaniem, niepowodzeniem zakończyły się próby identyfikacji genów różnicujących służących molekularnej klasyfikacji guzów Wilmsa, która byłaby skojarzona z klinicznym stadium choroby [99].
Z badań Takahashiego i wsp. [87] wynika, iż na postawie wzorca ekspresji można odróżnić guzy Wilmsa o dobrym rokowaniu, różniące się stopniem zaawansowania. W tej histologicznej grupie nerczaków wyniki leczenia są różne, a różnice w profilu ekspresji potwierdziły znaczną heterogenność w jej obrębie. Pomiędzy stadium I i II a III i IV stwierdzono różnice w poziomie ekspresji prawie 30 genów. Na podstawie analizy transkryptomu wywnioskowano również, iż podwyższenie poziomu topoizomerazy IIα oraz genów kodujących białka związane z mikrotubulami może wpływać na zwiększenie wrażliwości komórek nowotworowych na cytostatyki. Zirn i wsp. [106] stwierdzili istotne różnice w profilu ekspresji pomiędzy guzami Wilmsa z grup pośredniego i wysokiego ryzyka oraz dla nowotworów, w których dochodzi do wznowy. Jak zauważyli wspólny profil ekspresji miały, mimo różnic histologicznych, zarówno nowotwory należące do typu anaplastycznego, jak i z przewagą blastemy. Wyselekcjonowali zestaw 77 genów, służących odróżnieniu grupy nerczaków zarodkowych, w przypadku których może dojść do wznowy nowotworu.
Guzy kości (osteosarcoma, guz Ewinga)
Kostniakomięsak (osteosarcoma)
Kostniakomięsak (osteosarcoma) to najczęstszy typ nowotworu kości u dzieci, należący do stosunkowo opornych na leczenie [21]. Li i wsp. [52] przeanalizowali – za pomocą mikromacierzy cDNA – komórki nowotworowe pod kątem ich podatności na chemioterapię. Jak wskazują ich badania możliwe jest przeprowadzenie molekularnej klasyfikacji kostniakomięsaków na oporne i wrażliwe na leczenie już w chwili diagnozy. Podobnie Ochi i wsp. [66], na podstawie analizy ekspresji 60 genów, odróżnili dobrze od źle rokujących kostniakomięsaków. Jak wskazują badania przeprowadzone przez Mintza i wsp. [56], w powstanie zjawiska chemiooporności w omawianym typie nowotworów są zaangażowane 104 geny.
Mięsak Ewinga
Mięsak Ewinga (Ewing’s sarcoma – ES) jest drugim pod względem częstości występowania pierwotnym nowotworem kości [67]. Biologiczne i kliniczne czynniki prognostyczne są wciąż niewystarczające do klasyfikacji pacjentów do odpowiednich grup ryzyka. Ohali i wsp. [67] po przeanalizowaniu metodą macierzy genowych próbek pochodzących od pacjentów, u których stwierdzono nowotwór pierwotny lub występowanie przerzutu, otrzymali dwa odrębne profile ekspresji odpowiadające korzystnemu i złemu rokowaniu. Wykazano, że niekorzystne rokowanie i niepowodzenie terapii jest związane z nadekspresją genów regulujących cykl komórkowy: CDK2, ID1, CREB2, RAF 1, DMTF1, E2F3, E2F5; biorących udział w transdukcji sygnału: MAPK9, MKNK1, AXL, PDAP1, PTK7; czynników transkrypcyjnych: ZNF175, EP300, ZFP36L2, TAF6, TCFL4, EIF4A2; białek z rodziny RAS: RAB2, RAB1A, RHEB2; uczestniczących w adhezji komórkowej: CDH11, ITGAE, ITGB2; oraz związanych z inwazją i przerzutami: MTA1, HBXIP, kadheryna 11. Zmianom tym towarzyszy jednocześnie obniżenie liczby kopii transkryptów genów biorących udział w indukcji apoptozy: TNFRSF12, TGFB1, CASP10, TP63; naprawie DNA: IGHMBP2, XRCC2, ERCC2; histogenezie i organogenezie: PAX8, PAX6, COL6A1; formowaniu cytoszkieletu: BMP10, MYO1C, ARC; neurogenezie: NEUROD2, NTRK2, GRIN2A, SIM2; adhezji komórkowej: CDH2, ITGA2B, SCAM-1, ADAM15, ADAM19, ISLR; transdukcji sygnału: ESR2, ING1L, a także genów supresorowych: NEURL, FHIT, LLGL1, NF1 [67]. Jak podkreślili Ohali i wsp. wyniki analiz są w dużej mierze zgodne z otrzymanymi wcześniej przez Shipp i wsp. [83] dla DLBCL oraz przez Yeoha i wsp. [103] dla ALL. Istnienie wyraźnego wzorca ekspresji korelującego z rokowaniem już w czasie diagnozy potwierdza możliwość opracowania zindywidualizowanej strategii leczenia w oparciu o molekularną ocenę ryzyka również dla tego nowotworu [67].
Mięsaki
Mięsaki (sarcomas) są grupą nowotworów wywodzących się z tkanki łącznej (tkanka tłuszczowa, chrząstki, kości, mięśnie) lub jej prekursora – mezenchymy. Zalicza się do nich mięsaki tkanek miękkich (soft-tissue sarcomas – STS) oraz kostniakomięsaki [9,21]. W dość rzadkiej grupie chorób nowotworowych często spotyka się dużą różnorodność histologiczną oraz złożoność genetyczną, co stanowi utrudnienie klasycznej diagnozy [62]. Dlatego techniki analizy genetycznej są szansą na lepsze zrozumienie biologii tego rodzaju guzów [9]. Badania obejmujące grupę 16 klas mięsaków przeprowadzili Baird i wsp. [9]. Celem ich analiz było stworzenie uniwersalnych profili genetycznych, umożliwiających klasyfikację najczęstszych guzów litych dzieci i dorosłych. Metodą mikromacierzy ekspresyjnych zidentyfikowali geny, których ekpresja była najbardziej charakterystyczna dla każdej z analizowanych klas. Dla mięsaka Ewinga były to: FVT1, DCC, DKK2. JAK1; dla tłuszczakomięsaka: PPARG, FABP4, FALC5, SH3KBP1; dla miesaka gładkokomórkowego: MYLK, elementy kanałów sodowo- potasowych, CCN1, SLMAP; dla kostniakomięsaka: PPFIBP2, S100A13, PTHR1, P4HA2, PLOD, COL5A1, LUM, FGFR3, FGFR2, FGFR1; dla maziówczaka: FGF11, COL4A5, PBX3, BCE-1, TLE1, PDGFRA, EFNB3, NRP2, CXCL2, SHANK2, EGFR oraz mięsaka prążkowanokomórkowego (RMS): GAB1, MYCL1, MYOIE, IGF2, CCND2, PTPRF, PPFIA1, CDK6, FGFR4, GPC3, POU4F1.
Guzy drobnookrągłoniebieskokomórkowe (small round, blue cell tumors – SRBCTs)
Nowotwory drobnookrągłokomórkowe, takie jak: mięsak prążkowanokomórkowy, mięsak Ewinga, zwojak współczulny zarodkowy i chłoniaki złośliwe są podobne do siebie pod względem histologii (tabela 11). Khan i wsp. [43] przeanalizowali przydatność i dokładność wzorców ekspresji genów w klasyfikacji 4 typów nowotworów podobnych do siebie w rutynowej analizie histologicznej. Dzięki analizie danych w oparciu o sztuczne sieci neuronalne (artificial neuronal networks – ANNs) wykazali, iż jest możliwa identyfikacja charakterystycznych genów oraz poprawna klasyfikacja przypadków nerwiaków, guzów Ewinga, chłoniaków nieziarniczych oraz mięśniakomięsaków [43]. Badacze ci wyselekcjonowali 96 genów, których zróżnicowana ekspresja umożliwiła czułą i swoistą klasyfikację czterech analizowanych nowotworów.
Tabela 11. Guzy drobnookrągłoniebieskokomórkowe występujące u dzieci

Mięsaki tkanek miękkich (soft tissue sarcomas – STS)
Jest to liczna (50 podtypów) i zróżnicowana genetycznie oraz histologicznie grupa mięsaków. Dla większości złośliwych i łagodnych guzów litych tkanek miękkich (m.in. mięsaka Ewinga, maziówczaka, mięśniakomięsaka prążkowanokomórkowego, tłuszczakomięsaka) określono charakterystyczne liczbowe i strukturalne aberracje chromosomowe w komórkach nowotworowych [9,21].
Mięsak jasnokomórkowy (clear cell sarcoma – CCS)
Jest to rzadki nowotwór tkanek miękkich, stanowiący około 4% guzów nerek u dzieci [49,82]. CCS jest guzem, który trudno odróżnić od guzów złośliwych nerwów obwodowych (malignant peripheral nerve sheeth tumors – MPNST), pierwotnego czerniaka skóry typu guzowatego (primary nodular cutaneous melanoma), a także czerniaka skóry przerzutującego do tkanek miękkich (cutaneous melanoma metastatic to soft tissues) [82]. Segal i wsp. [82] zasugerowali, iż wprowadzenie do analizy profilu ekspresji genów może usprawnić klasyfikację tych nowotworów oraz sprecyzować dla nich kategorie diagnostyczne. Z badań wspomnianej grupy wynika, że mięsaka jasnokomórkowego można odróżnić od innych STS na podstawie danych o ekspresji, takich genów jak: CTNNA1, CDK2, CNP, SOX10, STC1, TCF8 oraz ERBB3/HER3. Genami różnicującymi CCS i czerniaki mogą być CREM, FGFR1 oraz IGFBP4, natomiast dane o poziomie ekspresji RABB33, MERKT, STC1 i GAL mogą być pomocne w odróżnieniu CCS zarówno od czerniaków jak i mięsaków tkanek miękkich. Ponadto Segal i wsp. [82] potwierdzili – na podstawie profilu ekspresji genów w CCS – wcześniejsze obserwacje, iż ten typ nowotworu jest genetycznym podtypem czerniaka, co ma znaczenie w zastosowaniu odpowiedniej terapii. Jednocześnie analizy profilu ekspresji umożliwiły identyfikację nowych potencjalnych cząsteczek docelowych dla leków (pMEL17, TYRP2/DCT, MELAN-A/MART-1, SOX10 oraz FGFR).
Mięśniakomięsak prążkowanokomórkowy (rhabdomyosarcoma – RMS)
Jest to najczęstszy bardzo złośliwy nowotwór tkanek miękkich występujący u dzieci [23,73]. Na podstawie morfologii wyróżnia się dwa typy RMS: zarodkowy (embryonal rhabdomyosarcoma – ERMS) oraz gorzej rokujący pęcherzykowy (alveolar rhabdomyosarcoma – ARMS) [95]. Prawdopodobnie w obrębie każdego podtypu możliwe jest dalsze wyodrębnienie podtypów jakimi są nowotwory z obecnością lub brakiem translokacji PAX3(7)-FKHR, o podobnych cechach histologicznych i immunohistochemicznych, ale odmiennych cechach klinicznych i złośliwości [21,23,95]. Wachtel i wsp. [95] stosując mikromacierz oligonukleotydową sklasyfikowali powyżej wymienione podtypy mięśniakomięsaków. Pomiędzy trzema analizowanymi grupami stwierdzono różnice w poziomie ekspresji kilkuset genów. W profilu ekspresji w komórkach z translokacją PAX3-FKHR obserwowano znaczne zwiększenie liczby transkryptów następujących genów: NCAM, MYCN, syntetazy argininobursztynianu, elastazy, ADAM10, AP2β oraz obniżenie liczby transkryptów genów: HOXC6, HOXC10, ZFH4, ZFP36. Pomiędzy ARMS pozytywnymi i negatywnymi pod względem obecności tej translokacji obserwowano również różnice w poziomach ekspresji genów receptorów. Nadekspresji następujących receptorów towarzyszyła obecność translokacji PAX3(7)-FKHR: receptor 1 kannabinoidów, FGFR2, FGFR4, receptor adrenergiczny α-2, receptor β3 acetylocholiny. W komórkach z brakiem PAX3-FKHR występował wysoki poziom ekspresji genów receptorów czynników wzrostu. Co więcej, na podstawie dalszych analiz zidentyfikowali nową translokację w przypadku tego nowotworu – PAX3/NCOA1 [95]. Badacze ci wykazali również, że identyfikacja trzech podtypów RMS jest możliwa dla kilku różnych zestawów genów i algorytmów analizy. Wachtel i wsp. [95] zauważyli również, że identyfikacja swoistych profili ekspresji genów przyspieszy i usprawni w przyszłości molekularną klasyfikację mięśniakomięsaków prążkowanych. Jest to szczególnie ważne w diagnostyce przypadków ARMS bez translokacji PAX3-FKHR, dla których nie ma jeszcze określonych wystarczających kryteriów histopatologicznych. Z kolei identyfikacja nowych ważnych genów, odgrywających rolę w rozwoju choroby nowotworowej otwiera drogę do opracowania nowych strategii terapeutycznych.
De Pitta i wsp. [23] wykorzystując tkankowoswoistą macierz cDNA przeprowadzili klasyfikację ARMS ze 100% dokładnością. Wykazali, że pomiędzy dwoma podtypami tego nowotworu występują zmiany w poziomach ekspresji 103 genów. Spośród tych genów wyselekcjonowali 4 geny: RAC1, CFL1, CCND1, IGFBP1, na podstawie których mogli odróżnić ARMS z translokacją PAX3-FKHR lub jej brakiem.
Guzy wątrobokomórkowe (hepatocellular carcinomas – HCC)
Nowotwory wywodzące się z hepatocytów również stanowią heterogenną grupę chorób. Przewidywanie ich przebiegu oraz dobór odpowiedniego leczenia wciąż stanowią problem. U dzieci najczęściej stwierdza się występowanie wątrobiaka zarodkowego (hepatoblastoma – HPBL) [55,102]. Zarodkowy guz wątroby jest trzecim co do częstości występowania złośliwym nowotworem wywodzącym się z narządów jamy brzusznej (po neuroblastoma i nephroblastoma) [105]. Pozytywnym rezultatem zakończono kilka projektów badawczych, których celem była molekularna charakterystyka, za pomocą profilu ekspresji genów, komórek patologicznych wątroby [1,38,60,102]. Udało się opracować molekularny system klasyfikacji, wyodrębnić geny, których zmiany ekspresji korelują z możliwością oceny prognozy przebiegu choroby oraz zidentyfikować nowe geny dla terapii celowanej [1,13,61]. Na uwagę zasługują badania przeprowadzone przez Luo i wsp. [55], w których wykazano wyraźne różnice na poziomie transkryptomu pomiędzy HCC, HPBL oraz prawidłową tkanką wątroby. HPBL w porównaniu z HCC charakteryzował się nadekspresją Mig6, TGFb1, DLK1, IGF2 oraz wyciszaniem ekspresji następujących genów: galektyny 4, ubikwityny 2 oraz adrenergicznego receptora α1.
W profilu ekspresji prawidłowych i patologicznych komórek odnotowano różnice w liczbie transkryptów 86 [102] lub 133 genów [60], ale nawet bardziej ograniczony zestaw najważniejszych genów okazał się wystarczający do scharakteryzowania komórek patologicznych. Model oparty na badaniu ekspresji 26 genów pozwalający na zdiagnozowanie (ze 100% dokładnością) wątrobiaka zarodkowego opracowali Nagata i wsp. [60]. Najważniejszymi genami różnicującymi komórki nowotworowe HPBL i komórki prawidłowe były: NAP1L1, STMN1, CCNG2, CDC7L1, IGF2, IGFBP4, ETV3, TPR, CD34 oraz NR1I3.
Wciąż brakuje analiz transkryptomu innych częstych nowotworów u dzieci, jakimi są retinoblastoma, histiocytoza z komórek Langerhansa oraz guzy zarodkowe.
PODSUMOWANIE
Diagnostyka i leczenie chorób nowotworowych u dzieci historycznie zostały oparte na rozpoznaniu histologicznym. Takie podejście okazało się niewystarczające, gdy morfologicznie podobne guzy wykazywały odmienny przebieg kliniczny. W licznych badaniach wykazano, iż technika mikromacierzy stanowi efektywne narzędzie molekularnej klasyfikacji najczęstszych nowotworów u dzieci, przewidywania przebiegu choroby, podziału grup ryzyka oraz identyfikacji genów do terapii celowanej. Połączenie profilowania ekspresji genów ze skutecznym algorytmem analizy danych staje się obiecującą metodą precyzyjnej i szybkiej diagnozy.
W przedstawionych przykładach analiz nowotworów u dzieci wykazano, iż stan genomu i transkryptomu charakteryzuje histologię komórek. Identyfikacja głównych genów i białek usprawnia poszukiwanie molekularnych markerów pomocnych w precyzyjnej klasyfikacji chorób nowotworowych. Badania molekularne wskazują, iż na podstawie odpowiedniego profilowania ekspresji genów możliwe jest przeprowadzenie właściwej stratyfikacji pacjentów do grup ryzyka, określenie ryzyka wznowy oraz wystąpienia przerzutu w chwili diagnozy. Kontynuacja takiego kierunku badań prawdopodobnie usprawni diagnostykę nowotworów hematologicznych i guzów litych oraz pozwoli na określenie roli poszczególnych cząsteczek w prognozowaniu klinicznym.
Jak wykazano w licznych badaniach głównymi zaletami molekularnej taksonomii są wysokie wartości czułości, swoistości i dokładności. Technika mikromacierzy oferuje szybką i dokładną analizę każdego typu komórek patologicznych. Określenie wzorców ekspresji może się okazać niezwykle pomocnym narzędziem klasyfikacji nowotworów podobnych do siebie pod względem histologicznym oraz tych nowotworów, które nie mają charakterystycznych rearanżacji genetycznych czy są wciąż słabo poznane. .
Stworzenie nowoczesnego systemu klasyfikacji w oparciu o molekularną charakterystykę komórek nowotworowych budzi nadzieje na opracowanie optymalnych strategii terapii dla indywidualnych pacjentów. Wyniki badań są w dużej mierze obiecujące. Wciąż istnieje potrzeba poszukiwania wzorców ekspresji korelujących z odpowiedzią na stosowane leczenie. Dużym wyzwaniem jest nadal optymalizacja i standaryzacja wykorzystywanych technik badawczych i algorytmów analizy danych.
PIŚMIENNICTWO
[1] Adesina A.M., Nguyen Y., Guanaratne P., Pulliam J., Lopez-Terrada D., Margolin J., Finegold M.: FOXG1 is overexpressed in hepatoblastoma. Hum. Pathol., 2007; 38: 400-409
[PubMed]
[2] Alizadeh A.A., Eisen M.B., Davis R.E., Ma C., Lossos I.S., Rosenwald A., Boldrick J.C., Sabet H., Tran T., Yu X., Powell J.I., Yang L., Marti G.E., Moore T., Hudson J.Jr., Lu L., Lewis D.B., Tibshirani R., Sherlock G., Chan W.C., Greiner T.C., Weisenburger D.D., Armitage J.O., Warnke R., Levy R., Wilson W., Grever M.R., Byrd J.C., Botstein D., Brown P.O., Staudt L.M.: Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature, 2000; 403: 503-511
[PubMed] [Full Text HTML] [Full Text PDF]
[3] Alizadeh A., Eisen M., Davis R.E., Ma C., Sabet H., Tran T., Powell J.I., Yang L., Marti G.E., Moore D.T., Hudson J.R.Jr., Chan W.C., Greiner T., Weisenburger D., Armitage J.O., Lossos I., Levy R., Botstein D., Brown P.O., Staudt L.M.: The lymphochip: a specialized cDNA microarray for the genomic-scale analysis of gene expression in normal and malignant lymphocytes. Cold Spring Harb. Symp. Quant. Biol., 1999; 64: 71-78
[PubMed]
[4] Andersson A., Eden P., Lindgren D., Nilsson J., Lassen C., Heldrup J., Fontes M., Borg A., Mitelman F., Johansson B., Hoglund M., Fioretos T.: Gene expression profiling of leukemic cell lines reveals conserved molecular signatures among subtypes with specific genetic aberrations. Leukemia, 2005; 19: 1042-1050
[PubMed]
[5] Andersson A., Olofsson T., Lindgren D., Nilsson B., Ritz C., Eden P., Lassen C., Rade J., Fontes M., Morse H., Heldrup J., Behrendtz M., Mitelman F., Hoglund M., Johansson B., Fioretos T.: Molecular signatures in childhood acute leukemia and their correlations to expression patterns in normal hematopoietic subpopulations. Proc. Natl. Acad. Sci. USA, 2005; 102: 19069-19074
[PubMed] [Full Text HTML] [Full Text PDF]
[6] Andersson A., Ritz C., Lindgren D., Eden P., Lassen C., Heldrup J., Olofsson T., Rade J., Fontes M., Porwit-Macdonald A., Behrendtz M., Hoglund M., Johansson B., Fioretos T.: Microarray-based classification of a consecutive series of 121 childhood acute leukemias: prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia, 2007; 21: 1198-1203
[PubMed]
[7] Armstrong S.A., Staunton J.E., Silverman L.B., Pieters R., den Boer M.L., Minden M.D., Sallan S.E., Lander E.S., Golub T.R., Korsmeyer S.J.: MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet., 2002; 30: 41-47
[PubMed] [Full Text HTML] [Full Text PDF]
[8] Bain B.: The WHO classification of the myelodysplastic syndromes. Exp. Oncol., 2004; 26: 166-169
[PubMed] [Full Text PDF]
[9] Baird K., Davis S., Antonescu C.R., Harper U.L., Walker R.L., Chen Y., Glatfelter A.A., Duray P.H., Meltzer P.S.: Gene expression profiling of human sarcomas: insights into sarcoma biology. Cancer Res., 2005; 65: 9226-9235
[PubMed] [Full Text HTML] [Full Text PDF]
[10] Belov L., de la Vega O., dos Remedios C.G., Mulligan S.P., Christopherson R.I.: Immunophenotyping of leukemias using a cluster of differentiation antibody microarray. Cancer Res., 2001; 61: 4483-4489
[PubMed] [Full Text HTML] [Full Text PDF]
[11] Belov L., Huang P., Barber N., Mulligan S.P., Christopherson R.I.: Identification of repertoires of surface antigens on leukemias using an antibody microarray. Proteomics, 2003; 3: 2147-2154
[PubMed]
[12] Belov L., Mulligan S.P., Barber N., Woolfson A., Scott M., Stoner K., Chrisp J.S., Sewell W.A., Bradstock K.F., Bendall L., Pascovici D.S., Thomas M., Erber W., Huang P., Sartor M., Young G.A., Wiley J.S., Juneja S., Wierda W.G., Green A.R., Keating M.J., Christopherson R.I.: Analysis of human leukaemias and lymphomas using extensive immunophenotypes from an antibody microarray. Br. J. Haematol., 2006; 135: 184-197
[PubMed]
[13] Breuhahn K., Vreden S., Haddad R., Beckebaum S., Stippel D., Flemming P., Nussbaum T., Caselmann W.H., Haab B.B., Schirmacher P.: Molecular profiling of human hepatocellular carcinoma defines mutually exclusive interferon regulation and insulin-like growth factor II overexpression. Cancer Res., 2004; 64: 6058-6064
[PubMed] [Full Text HTML] [Full Text PDF]
[14] Bullinger L., Dohner K., Bair E., Frohling S., Schlenk R.F., Tibshirani R., Dohner H., Pollack J.R.: Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N. Engl. J. Med., 2004; 350: 1605-1616
[PubMed] [Full Text HTML] [Full Text PDF]
[15] Bullinger L., Doner H., Pollack J.R.: Genomics in myeloid leukemias: an array of possibilities. Rev. Clin. Exp. Hematol., 2005; 9: E2
[PubMed]
[16] Bullinger L., Valk P.J.: Gene expression profiling in acute myeloid leukemia. J. Clin. Oncol., 2005; 23: 6296-6305
[PubMed]
[17] Cario G., Stanulla M., Fine B.M., Teuffel O., Neuhoff N.V., Schrauder A., Flohr T., Schafer B.W., Bartram C.R., Welte K., Schlegelberger B., Schrappe M.: Distinct gene expression profiles determine molecular treatment response in childhood acute lymphoblastic leukemia. Blood, 2005; 105: 821-826
[PubMed] [Full Text HTML] [Full Text PDF]
[18] Catalano A., Iland H.: Molecular biology of lymphoma in the microarray era. Pathology, 2005; 37: 508-522
[PubMed]
[19] Chung C.H., Levy S., Chaurand P., Carbone D.P.: Genomics and proteomics: emerging technologies in clinical cancer research. Crit. Rev. Oncol. Hematol., 2007; 61: 1-25
[PubMed]
[20] Chybicka A.: Najnowsze osiągnięcia w onkologii dziecięcej. Przew. Lek., 2007; 2: 174-180
[21] Dang T.A., Man T.K.: Classification of sarcomas using bioinformatics and molecular profiling. Curr. Pharm. Biotechnol., 2007; 8: 83-91
[PubMed]
[22] Dave S.S., Fu K., Wright G.W., Lam L.T., Kluin P., Boerma E.J., Greiner T.C., Weisenburger D.D., Rosenwald A., Ott G., Muller-Hermelink H.K., Gascoyne R.D., Delabie J., Rimsza L.M., Braziel R.M., Grogan T.M., Campo E., Jaffe E.S., Dave B.J., Sanger W., Bast M., Vose J.M., Armitage J.O., Connors J.M., Smeland E.B., Kvaloy S., Holte H., Fisher R.I., Miller T.P., Montserrat E., Wilson W.H., Bahl M., Zhao H., Yang L., Powell J., Simon R., Chan W.C., Staudt L.M.: Molecular diagnosis of Burkitt’s lymphoma. N. Engl. J. Med., 2006; 354: 2431-2442
[PubMed] [Full Text HTML] [Full Text PDF]
[23] De Pitta C., Tombolan L., Albiero G., Sartori F., Romualdi C., Jurman G., Carli M., Furlanello C., Lanfranchi G., Rosolen A.: Gene expression profiling identifies potential relevant genes in alveolar rhabdomyosarcoma pathogenesis and discriminates PAX3-FKHR positive and negative tumors. Int. J. Cancer, 2006; 118: 2772-2781
[PubMed]
[24] Debernardi S., Lillington D.M., Chaplin T., Tomlinson S., Amess J., Rohatiner A., Lister T.A., Young B.D.: Genome-wide analysis of acute myeloid leukemia with normal karyotype reveals a unique pattern of homeobox gene expression distinct from those with translocation-mediated fusion events. Genes Chromosomes Cancer, 2003; 37: 149-158
[PubMed]
[25] Devilard E., Bertucci F., Trempat P., Bouabdallah R., Loriod B., Giaconia A., Brousset P., Granjeaud S., Nguyen C., Birnbaum D., Birg F., Houlgatte R., Xerri L.: Gene expression profiling defines molecular subtypes of classical Hodgkin’s disease. Oncogene, 2002; 21: 3095-3102
[PubMed] [Full Text HTML] [Full Text PDF]
[26] Dmoszyńska A., Robak, T. (red.).: Podstawy hematologii. Lublin: CZELEJ; 2003.
[27] Edick M.J., Cheng C., Yang W., Cheok M., Wilkinson M.R., Pei D., Evans W.E., Kun L.E., Pui C.H., Relling M.V.: Lymphoid gene expression as a predictor of risk of secondary brain tumors. Genes Chromosomes Cancer, 2005; 42: 107-116
[PubMed]
[28] Ferrando A.A., Neuberg D.S., Staunton J., Loh M.L., Huard C., Raimondi S.C., Behm F.G., Pui C.H., Downing J.R., Gilliland D.G., Lander E.S., Golub T.R., Look A.T.: Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell, 2002; 1: 75-87
[PubMed] [Full Text HTML] [Full Text PDF]
[29] Fine B.M., Stanulla M., Schrappe M., Ho M., Viehmann S., Harbott J., Boxer L.M.: Gene expression patterns associated with recurrent chromosomal translocations in acute lymphoblastic leukemia. Blood, 2004; 103: 1043-1049
[PubMed] [Full Text HTML] [Full Text PDF]
[30] Ghobrial I.M., McCormick D.J., Kaufmann S.H., Leontovich A.A., Loegering D.A., Dai N.T., Krajnik K.L., Stenson M.J., Melhem M.F., Novak A.J., Ansell S.M., Witzig T.E.: Proteomic analysis of mantle-cell lymphoma by protein microarray. Blood, 2005; 105: 3722-3730
[PubMed] [Full Text HTML] [Full Text PDF]
[31] Golub T.R., Slonim D.K., Tamayo P., Huard C., Gaasenbeek M., Mesirov J.P., Coller H., Loh M.L., Downing J.R., Caligiuri M.A., Bloomfield C.D., Lander E.S.: Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science, 1999; 286: 531-537
[PubMed]
[32] Gottardo N.G., Hoffmann K., Beesley A.H., Freitas J.R., Firth M.J., Perera K.U., de Klerk N.H., Baker D.L., Kees U.R.: Identification of novel molecular prognostic markers for paediatric T-cell acute lymphoblastic leukaemia. Br. J. Haematol., 2007; 137: 319-328
[PubMed]
[33] Grimwade D., Haferlach T.: Gene-expression profiling in acute myeloid leukemia. N. Engl. J. Med., 2004; 350: 1676-1678
[PubMed]
[34] Haferlach T., Kohlmann A., Kern W., Hiddemann W., Schnittger S., Schoch C.: Gene expression profiling as a tool for the diagnosis of acute leukemias. Semin. Hematol., 2003; 40: 281-295
[PubMed]
[35] Haferlach T., Kohlmann A., Schnittger S., Dugas M., Hiddemann W., Kern W., Schoch C.: Global approach to the diagnosis of leukemia using gene expression profiling. Blood, 2005; 106: 1189-1198
[PubMed] [Full Text HTML] [Full Text PDF]
[36] Holleman A., Cheok M.H., den Boer M.L., Yang W., Veerman A.J., Kazemier K.M., Pei D., Cheng C., Pui C.H., Relling M.V., Janka-Schaub G.E., Pieters R., Evans W.E.: Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N. Engl. J. Med., 2004; 351: 533-542
[PubMed] [Full Text HTML] [Full Text PDF]
[37] Holleman A., den Boer M.L., de Menezes R.X., Cheok M.H., Cheng C., Kazemier K.M., Janka-Schaub G.E., Gobel U., Graubner U.B., Evans W.E., Pieters R.: The expression of 70 apoptosis genes in relation to lineage, genetic subtype, cellular drug resistance, and outcome in childhood acute lymphoblastic leukemia. Blood, 2006; 107: 769-776
[PubMed] [Full Text HTML] [Full Text PDF]
[38] Janoueix-Lerosey I., Novikov E., Monteiro M., Gruel N., Schleiermacher G., Loriod B., Nguyen C., Delattre O.: Gene expression profiling of 1p35-36 genes in neuroblastoma. Oncogene, 2004; 23: 5912-5922
[PubMed]
[39] Jemal A., Siegel R., Ward E., Murray T., Xu J., Thun M.J.: Cancer statistics, 2007. CA Cancer J. Clin., 2007; 57: 43-66
[PubMed] [Full Text HTML] [Full Text PDF]
[40] Kaspers G.J., Zwaan C.M.: Pediatric acute myeloid leukemia: towards high-quality cure of all patients. Haematologica, 2007; 92: 1519-1532
[PubMed] [Full Text HTML] [Full Text PDF]
[41] Kees U.R.: Gene expression signatures in lymphoid tumours. Immunol. Cell Biol., 2004; 82: 154-160
[PubMed]
[42] Kern W., Kohlmann A., Wuchter C., Schnittger S., Schoch C., Mergenthaler S., Ratei R., Ludwig W.D., Hiddemann W., Haferlach T.: Correlation of protein expression and gene expression in acute leukemia. Cytometry B. Clin. Cytom., 2003; 55: 29-36
[PubMed] [Full Text HTML] [Full Text PDF]
[43] Khan J., Wei J.S., Ringner M., Saal L.H., Ladanyi M., Westermann F., Berthold F., Schwab M., Antonescu C.R., Peterson C., Meltzer P.S.: Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat. Med., 2001; 7: 673-679
[PubMed]
[44] Kohlmann A., Schoch C., Schnittger S., Dugas M., Hiddemann W., Kern W., Haferlach T.: Molecular characterization of acute leukemias by use of microarray technology. Genes Chromosomes Cancer, 2003; 37: 396-405
[PubMed]
[45] Kohlmann A., Schoch C., Schnittger S., Dugas M., Hiddemann W., Kern W., Haferlach T.: Pediatric acute lymphoblastic leukemia (ALL) gene expression signatures classify an independent cohort of adult ALL patients. Leukemia, 2004; 18: 63-71
[PubMed]
[46] Korshunov A., Neben K., Wrobel G., Tews B., Benner A., Hahn M., Golanov A., Lichter P.: Gene expression patterns in ependymomas correlate with tumor location, grade, and patient age. Am. J. Pathol., 2003; 163: 1721-1727
[PubMed] [Full Text HTML] [Full Text PDF]
[47] Kowal M., Wiliński G.: Wybrane aspekty biologii choroby Hodgkina w świetle ostatnich badań. Onkol. Pol., 2000; 3: 109-115
[Full Text PDF]
[48] Kowalczyk J.R., Dudkiewicz E., Balwierz W., Boguslawska-Jaworska J., Rokicka-Milewska R.: Incidence of childhood cancers in Poland in 1995-1999. Med. Sci. Monit., 2002; 8: CR587-CR590
[PubMed]
[49] Kowalski J., Madziara W.: Nowotwory nerek wieku dziecięcego. Przew. Lek., 2005; 9: 76-82
[50] Krasnoselsky A.L., Whiteford C.C., Wei J.S., Bilke S., Westermann F., Chen Q.R., Khan J.: Altered expression of cell cycle genes distinguishes aggressive neuroblastoma. Oncogene, 2005; 24: 1533-1541
[PubMed]
[51] Lacayo N.J., Meshinchi S., Kinnunen P., Yu R., Wang Y., Stuber C.M., Douglas L., Wahab R., Becton D.L., Weinstein H., Chang M.N., Willman C.L., Radich J.P., Tibshirani R., Ravindranath Y., Sikic B.I., Dahl G.V.: Gene expression profiles at diagnosis in de novo childhood AML patients identify FLT3 mutations with good clinical outcomes. Blood, 2004; 104: 2646-2654
[PubMed] [Full Text HTML] [Full Text PDF]
[52] Li Y., Dang T.A., Shen J., Perlaky L., Hicks J., Murray J., Meyer W., Chintagumpala M., Lau C.C., Man T.K.: Identification of a plasma proteomic signature to distinguish pediatric osteosarcoma from benign osteochondroma. Proteomics, 2006; 6: 3426-3435
[PubMed]
[53] Liu E.T.: Classification of cancers by expression profiling. Curr. Opin. Genet. Dev., 2003; 13: 97-103
[PubMed]
[54] Lugthart S., Cheok M.H., den Boer M.L., Yang W., Holleman A., Cheng C., Pui C.H., Relling M.V., Janka-Schaub G.E., Pieters R., Evans W.E.: Identification of genes associated with chemotherapy crossresistance and treatment response in childhood acute lymphoblastic leukemia. Cancer Cell, 2005; 7: 375-386
[PubMed] [Full Text PDF]
[55] Luo J.H., Ren B., Keryanov S., Tseng G.C., Rao U.N., Monga S.P., Strom S., Demetris A.J., Nalesnik M., Yu Y.P., Ranganathan S., Michalopoulos G.K.: Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology, 2006; 44: 1012-1024
[PubMed] [Full Text HTML] [Full Text PDF]
[56] Mintz M.B., Sowers R., Brown K.M., Hilmer S.C., Mazza B., Huvos A.G., Meyers P.A., Lafleur B., McDonough W.S., Henry M.M., Ramsey K.E., Antonescu C.R., Chen W., Healey J.H., Daluski A., Berens M.E., Macdonald T.J., Gorlick R., Stephan D.A.: An expression signature classifies chemotherapy-resistant pediatric osteosarcoma. Cancer Res., 2005; 65: 1748-1754
[PubMed] [Full Text HTML] [Full Text PDF]
[57] Moos P.J., Raetz E.A., Carlson M.A., Szabo A., Smith F.E., Willman C., Wei Q., Hunger S.P., Carroll W.L.: Identification of gene expression profiles that segregate patients with childhood leukemia. Clin. Cancer Res., 2002; 8: 3118-3130
[PubMed] [Full Text HTML] [Full Text PDF]
[58] Mrozek K., Marcucci G., Paschka P., Whitman S.P., Bloomfield C.D.: Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood, 2007; 109: 431-448
[PubMed] [Full Text HTML] [Full Text PDF]
[59] Mullighan C.G., Flotho C., Downing J.R.: Genomic assessment of pediatric acute leukemia. Cancer J., 2005; 11: 268-282
[PubMed]
[60] Nagata T., Takahashi Y., Ishii Y., Asai S., Nishida Y., Murata A., Koshinaga T., Fukuzawa M., Hamazaki M., Asami K., Ito E., Ikeda H., Takamatsu H., Koike K., Kikuta A., Kuroiwa M., Watanabe A., Kosaka Y., Fujita H., Miyake M., Mugishima H.: Transcriptional profiling in hepatoblastomas using high-density oligonucleotide DNA array. Cancer Genet. Cytogenet., 2003; 145: 152-160
[PubMed]
[61] Nam S.W., Park J.Y., Ramasamy A., Shevade S., Islam A., Long P.M., Park C.K., Park S.E., Kim S.Y., Lee S.H., Park W.S., Yoo N.J., Liu E.T., Miller L.D., Lee J.Y.: Molecular changes from dysplastic nodule to hepatocellular carcinoma through gene expression profiling. Hepatology, 2005; 42: 809-818
[PubMed] [Full Text HTML] [Full Text PDF]
[62] Nielsen T.O., West R.B., Linn S.C., Alter O., Knowling M.A., O’Connell J.X., Zhu S., Fero M., Sherlock G., Pollack J.R., Brown P.O., Botstein D., van de Rijn M.: Molecular characterisation of soft tissue tumours: a gene expression study. Lancet, 2002; 359: 1301-1307
[PubMed]
[63] Nilsson B., Andersson A., Johansson M., Fioretos T.: Cross-platform classification in microarray-based leukemia diagnostics. Haematologica, 2006; 91: 821-824
[PubMed] [Full Text PDF]
[64] Oberthuer A., Berthold F., Warnat P., Hero B., Kahlert Y., Spitz R., Ernestus K., Konig R., Haas S., Eils R., Schwab M., Brors B., Westermann F., Fischer M.: Customized oligonucleotide microarray gene expression-based classification of neuroblastoma patients outperforms current clinical risk stratification. J. Clin. Oncol., 2006; 24: 5070-5078
[PubMed] [Full Text PDF]
[65] Oberthuer A., Warnat P., Kahlert Y., Westermann F., Spitz R., Brors B., Hero B., Eils R., Schwab M., Berthold F., Fischer M.: Classification of neuroblastoma patients by published gene-expression markers reveals a low sensitivity for unfavorable courses of MYCN non-amplified disease. Cancer Lett., 2007; 250: 250-267
[PubMed]
[66] Ochi K., Daigo Y., Katagiri T., Nagayama S., Tsunoda T., Myoui A., Naka N., Araki N., Kudawara I., Ieguchi M., Toyama Y., Toguchida J., Yoshikawa H., Nakamura Y.: Prediction of response to neoadjuvant chemotherapy for osteosarcoma by gene-expression profiles. Int. J. Oncol., 2004; 24: 647-655
[PubMed]
[67] Ohali A., Avigad S., Zaizov R., Ophir R., Horn-Saban S., Cohen I.J., Meller I., Kollender Y., Issakov J., Yaniv I.: Prediction of high risk Ewing’s sarcoma by gene expression profiling. Oncogene, 2004; 23: 8997-9006
[PubMed]
[68] Ozols R.F., Herbst R.S., Colson Y.L., Gralow J., Bonner J., Curran W.J.Jr., Eisenberg B.L., Ganz P.A., Kramer B.S., Kris M.G., Markman M., Mayer R.J., Raghavan D., Reaman G.H., Sawaya R., Schilsky R.L., Schuchter L.M., Sweetenham J.W., Vahdat L.T., Winn R.J.: Clinical cancer advances 2006: major research advances in cancer treatment, prevention, and screening – a report from the American Society of Clinical Oncology. J. Clin. Oncol., 2007; 25: 146-162
[PubMed] [Full Text PDF]
[69] Pomeroy S.L., Tamayo P., Gaasenbeek M., Sturla L.M., Angelo M., McLaughlin M.E., Kim J.Y., Goumnerova L.C., Black P.M., Lau C., Allen J.C., Zagzag D., Olson J.M., Curran T., Wetmore C., Biegel J.A., Poggio T., Mukherjee S., Rifkin R., Califano A., Stolovitzky G., Louis D.N., Mesirov J.P., Lander E.S., Golub T.R.: Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature, 2002; 415: 436-442
[PubMed]
[70] Ries L.A.G., Smith, M.A., Gurney, J.G. Linet, M., Tamra, T., Young, J.L., Bunin, G.R. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda: National Cancer Institute, SEER Program. NIH Pub. No. 99-4649; 1999
[71] Ring B.Z., Ross D.T.: Microarrays and molecular markers for tumor classification. Genome Biol., 2002; 3: comment2005
[PubMed] [Full Text HTML] [Full Text PDF]
[72] Roela R.A., Carraro D.M., Brentani H.P., Kaiano J.H., Simao D.F., Guarnieiro R., Lopes L.F., Borojevic R., Brentani M.M.: Gene stage-specific expression in the microenvironment of pediatric myelodysplastic syndromes. Leuk. Res., 2007; 31: 579-589
[PubMed]
[73] Romualdi C., De Pitta C., Tombolan L., Bortoluzzi S., Sartori F., Rosolen A., Lanfranchi G.: Defining the gene expression signature of rhabdomyosarcoma by meta-analysis. BMC Genomics, 2006; 7: 287
[PubMed] [Full Text HTML] [Full Text PDF]
[74] Rosenwald A., Staudt L.M.: Gene expression profiling of diffuse large B-cell lymphoma. Leuk. Lymphoma, 2003; 44 Suppl 3: S41-S47
[PubMed]
[75] Rosenwald A., Wright G., Chan W.C., Connors J.M., Campo E., Fisher R.I., Gascoyne R.D., Muller-Hermelink H.K., Smeland E.B., Giltnane J.M., Hurt E.M., Zhao H., Averett L., Yang L., Wilson W.H., Jaffe E.S., Simon R., Klausner R.D., Powell J., Duffey P.L., Longo D.L., Greiner T.C., Weisenburger D.D., Sanger W.G., Dave B.J., Lynch J.C., Vose J., Armitage J.O., Montserrat E., Lopez-Guillermo A., Grogan T.M., Miller T.P., LeBlanc M., Ott G., Kvaloy S., Delabie J., Holte H., Krajci P., Stokke T., Staudt L.M.: The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med., 2002; 346: 1937-1947
[PubMed] [Full Text HTML] [Full Text PDF]
[76] Ross M.E., Mahfouz R., Onciu M., Liu H.C., Zhou X., Song G., Shurtleff S.A., Pounds S., Cheng C., Ma J., Ribeiro R.C., Rubnitz J.E., Girtman K., Williams W.K., Raimondi S.C., Liang D.C., Shih L.Y., Pui C.H., Downing J.R.: Gene expression profiling of pediatric acute myelogenous leukemia. Blood, 2004; 104: 3679-3687
[PubMed] [Full Text HTML] [Full Text PDF]
[77] Ross M.E., Zhou X., Song G., Shurtleff S.A., Girtman K., Williams W.K., Liu H.C., Mahfouz R., Raimondi S.C., Lenny N., Patel A., Downing J.R.: Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood, 2003; 102: 2951-2959
[PubMed] [Full Text HTML] [Full Text PDF]
[78] Roszkowski M., Grajkowska W.: Postępy w poznaniu biologii molekularnej guzów zarodkowych OUN i możliwości planowania molekularnej terapii celowanej. Neurol. Neurochir. Pol., 2006; 40: 212-218
[PubMed]
[79] Schoch C., Kohlmann A., Schnittger S., Brors B., Dugas M., Mergenthaler S., Kern W., Hiddemann W., Eils R., Haferlach T.: Acute myeloid leukemias with reciprocal rearrangements can be distinguished by specific gene expression profiles. Proc. Natl. Acad. Sci. USA, 2002; 99: 10008-10013
[PubMed] [Full Text HTML] [Full Text PDF]
[80] Schramm A., Schulte J.H., Klein-Hitpass L., Havers W., Sieverts H., Berwanger B., Christiansen H., Warnat P., Brors B., Eils J., Eils R., Eggert A.: Prediction of clinical outcome and biological characterization of neuroblastoma by expression profiling. Oncogene, 2005; 24: 7902-7912
[PubMed]
[81] Scotting P.J., Walker D.A., Perilongo G.: Childhood solid tumours: a developmental disorder. Nat. Rev. Cancer, 2005; 5: 481-488
[PubMed]
[82] Segal N.H., Pavlidis P., Noble W.S., Antonescu C.R., Viale A., Wesley U.V., Busam K., Gallardo H., DeSantis D., Brennan M.F., Cordon-Cardo C., Wolchok J.D., Houghton A.N.: Classification of clear-cell sarcoma as a subtype of melanoma by genomic profiling. J. Clin. Oncol., 2003; 21: 1775-1781
[PubMed] [Full Text PDF]
[83] Shipp M.A., Ross K.N., Tamayo P., Weng A.P., Kutok J.L., Aguiar R.C., Gaasenbeek M., Angelo M., Reich M., Pinkus G.S., Ray T.S., Koval M.A., Last K.W., Norton A., Lister T.A., Mesirov J., Neuberg D.S., Lander E.S., Aster J.C., Golub T.R.: Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat. Med., 2002; 8: 68-74
[PubMed] [Full Text HTML] [Full Text PDF]
[84] Staal F.J., van der Burg M., Wessels L.F., Barendregt B.H., Baert M.R., van den Burg C.M., van Huffel C., Langerak A.W., van der Velden V.H., Reinders M.J., van Dongen J.J.: DNA microarrays for comparison of gene expression profiles between diagnosis and relapse in precursor-B acute lymphoblastic leukemia: choice of technique and purification influence the identification of potential diagnostic markers. Leukemia, 2003; 17: 1324-1332
[PubMed]
[85] Steliarova-Foucher E., Stiller C., Lacour B., Kaatsch P.: International Classification of Childhood Cancer, third edition. Cancer, 2005; 103: 1457-1467
[PubMed] [Full Text HTML] [Full Text PDF]
[86] Szczepanek J., Styczynski J., Haus O., Tretyn A., Wysocki M.: Profile ekspresji genów w ostrej białaczce limfoblastycznej u dzieci i dorosłych. Post. Hig. Med. Dośw., 2007; 61: 519-533
[PubMed] [Full Text HTML] [Full Text PDF]
[87] Takahashi M., Yang X.J., Lavery T.T., Furge K.A., Williams B.O., Tretiakova M., Montag A., Vogelzang N.J., Re G.G., Garvin A.J., Soderhall S., Kagawa S., Hazel-Martin D., Nordenskjold A., Teh B.T.: Gene expression profiling of favorable histology Wilms tumors and its correlation with clinical features. Cancer Res., 2002; 62: 6598-6605
[PubMed] [Full Text HTML] [Full Text PDF]
[88] Taylor M.D., Poppleton H., Fuller C., Su X., Liu Y., Jensen P., Magdaleno S., Dalton J., Calabrese C., Board J., Macdonald T., Rutka J., Guha A., Gajjar A., Curran T., Gilbertson R.J.: Radial glia cells are candidate stem cells of ependymoma. Cancer Cell, 2005; 8: 323-335
[PubMed] [Full Text HTML] [Full Text PDF]
[89] Teuffel O., Dettling M., Cario G., Stanulla M., Schrappe M., Buhlmann P., Niggli F.K., Schafer B.W.: Gene expression profiles and risk stratification in childhood acute lymphoblastic leukemia. Haematologica, 2004; 89: 801-808
[PubMed] [Full Text PDF]
[90] Thorgeirsson S.S., Lee J.S., Grisham J.W.: Molecular prognostication of liver cancer: end of the beginning. J. Hepatol., 2006; 44: 798-805
[PubMed]
[91] Tsai C.A., Chen C.H., Lee T.C., Ho I.C., Yang U.C., Chen J.J.: Gene selection for sample classifications in microarray experiments. DNA Cell Biol., 2004; 23: 607-614
[PubMed]
[92] Valk P.J., Delwel R., Lowenberg B.: Gene expression profiling in acute myeloid leukemia. Curr. Opin. Hematol., 2005; 12: 76-81
[PubMed]
[93] Valk P.J., Verhaak R.G., Beijen M.A., Erpelinck C.A., Barjesteh van Waalwijk van Doorn-Khosrovani S., Boer J.M., Beverloo H.B., Moorhouse M.J., van der Spek P.J., Lowenberg B., Delwel R.: Prognostically useful gene-expression profiles in acute myeloid leukemia. N. Engl. J. Med., 2004; 350: 1617-1628
[PubMed] [Full Text HTML] [Full Text PDF]
[94] Virtaneva K., Wright F.A., Tanner S.M., Yuan B., Lemon W.J., Caligiuri M.A., Bloomfield C.D., de La Chapelle A., Krahe R.: Expression profiling reveals fundamental biological differences in acute myeloid leukemia with isolated trisomy 8 and normal cytogenetics. Proc. Natl. Acad. Sci. USA, 2001; 98: 1124-1129
[PubMed] [Full Text HTML] [Full Text PDF]
[95] Wachtel M., Dettling M., Koscielniak E., Stegmaier S., Treuner J., Simon-Klingenstein K., Buhlmann P., Niggli F.K., Schafer B.W.: Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res., 2004; 64: 5539-5545
[PubMed] [Full Text HTML] [Full Text PDF]
[96] Warnat P., Oberthuer A., Fischer M., Westermann F., Eils R., Brors B.: Cross-study analysis of gene expression data for intermediate neuroblastoma identifies two biological subtypes. BMC Cancer, 2007; 7: 89
[PubMed] [Full Text HTML] [Full Text PDF]
[97] Wei J.S., Greer B.T., Westermann F., Steinberg S.M., Son C.G., Chen Q.R., Whiteford C.C., Bilke S., Krasnoselsky A.L., Cenacchi N., Catchpoole D., Berthold F., Schwab M., Khan J.: Prediction of clinical outcome using gene expression profiling and artificial neural networks for patients with neuroblastoma. Cancer Res., 2004; 64: 6883-6891
[PubMed] [Full Text HTML] [Full Text PDF]
[98] Willenbrock H., Juncker A.S., Schmiegelow K., Knudsen S., Ryder L.P.: Prediction of immunophenotype, treatment response, and relapse in childhood acute lymphoblastic leukemia using DNA microarrays. Leukemia, 2004; 18: 1270-1277
[PubMed]
[99] Williams R.D., Hing S.N., Greer B.T., Whiteford C.C., Wei J.S., Natrajan R., Kelsey A., Rogers S., Campbell C., Pritchard-Jones K., Khan J.: Prognostic classification of relapsing favorable histology Wilms tumor using cDNA microarray expression profiling and support vector machines. Genes Chromosomes Cancer, 2004; 41: 65-79
[PubMed]
[100] Willman C.L.: Discovery of novel molecular classification schemes and genes predictive of outcome in leukemia. Hematol. J., 2004; 5 Suppl 3: S138-S143
[PubMed]
[101] Yagi T., Morimoto A., Eguchi M., Hibi S., Sako M., Ishii E., Mizutani S., Imashuku S., Ohki M., Ichikawa H.: Identification of a gene expression signature associated with pediatric AML prognosis. Blood, 2003; 102: 1849-1856
[PubMed] [Full Text HTML] [Full Text PDF]
[102] Yamada S., Ohira M., Horie H., Ando K., Takayasu H., Suzuki Y., Sugano S., Hirata T., Goto T., Matsunaga T., Hiyama E., Hayashi Y., Ando H., Suita S., Kaneko M., Sasaki F., Hashizume K., Ohnuma N., Nakagawara A.: Expression profiling and differential screening between hepatoblastomas and the corresponding normal livers: identification of high expression of the PLK1 oncogene as a poor-prognostic indicator of hepatoblastomas. Oncogene, 2004; 23: 5901-5911
[PubMed]
[103] Yeoh E.J., Ross M.E., Shurtleff S.A., Williams W.K., Patel D., Mahfouz R., Behm F.G., Raimondi S.C., Relling M.V., Patel A., Cheng C., Campana D., Wilkins D., Zhou X., Li J., Liu H., Pui C.H., Evans W.E., Naeve C., Wong L., Downing J.R.: Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell, 2002; 1: 133-143
[PubMed] [Full Text HTML] [Full Text PDF]
[104] Zhou X., Rao N.P., Cole S.W., Mok S.C., Chen Z., Wong D.T.: Progress in concurrent analysis of loss of heterozygosity and comparative genomic hybridization utilizing high density single nucleotide polymorphism arrays. Cancer Genet. Cytogenet., 2005; 159: 53-57
[PubMed]
[105] Zielińska E., Misiak G., Wegner O., Bodalski J.: Trudności diagnostyczne i terapeutyczne zaawansowanych stadiów hepatoblastoma – doświadczenia własne i przegląd piśmiennictwa. Przegl. Pediatr., 2004; 34: 254-257
[Abstract] [Full Text PDF]
[106] Zirn B., Hartmann O., Samans B., Krause M., Wittmann S., Mertens F., Graf N., Eilers M., Gessler M.: Expression profiling of Wilms tumors reveals new candidate genes for different clinical parameters. Int. J. Cancer, 2006; 118: 1954-1962
[PubMed]
[107] Zirn B., Samans B., Spangenberg C., Graf N., Eilers M., Gessler M.: All-trans retinoic acid treatment of Wilms tumor cells reverses expression of genes associated with high risk and relapse in vivo. Oncogene, 2005; 24: 5246-5251
[PubMed]