
Molekularne mechanizmy aktywności przeciwnowotworowej taksanów. I. Oddziaływanie docetakselu na mikrotubule
Sabina Tabaczar 1 , Aneta Koceva-Chyła 2 , Karolina Matczak 2 , Krzysztof Gwoździński 1Streszczenie
Docetaksel (Taxotere) jest nowym, półsyntetycznym taksoidem, otrzymywanym z 10-deacetylobakatyny, naturalnej substancji, uzyskiwanej z igieł cisu pospolitego (Taxus baccata). Lek należy do grupy związków będących inhibitorami mitozy i jest szeroko stosowany zarówno w monoterapii, jak i w połączeniu z innymi lekami przeciwnowotworowymi, w różnych typach nowotworów. W artykule przedstawiono istniejące schematy chemioterapii z zastosowaniem docetakselu, jego farmakokinetykę i metabolizm. Omówiono budowę, dynamikę i funkcję mikrotubul, które są głównym celem działania docetakselu w komórce. Mikrotubule są dynamicznymi strukturami, zbudowanymi z heterodimerów a- i b-tubuliny. Ich zachowanie się w komórce nosi nazwę dynamicznej niestabilności. Proces ten związany jest ze zdolnością tubuliny do hydrolizowania GTP. Hydroliza GTP do GDP powoduje odłączenie się heterodimerów α- i β-tubuliny (depolimeryzację) i skrócenie mikrotubuli. Przyłączenie do mikrotubul heterodimerów α- i β-tubuliny ze związanym GTP prowadzi natomiast do ich polimeryzacji i wydłużenia. Procesy polimeryzacji i depolimeryzacji mikrotubul mają podstawowe znaczenie dla podziału mitotycznego komórki. Docetaksel łączy się z β-tubuliną, przez co hamuje depolimeryzację mikrotubul i stabilizuje je. Naruszenie dynamicznej niestabilności mikrotubul powoduje zatrzymanie komórek w mitozie, na granicy metafazy i anafazy, co prowadzi do apoptozy lub innego typu śmierci komórkowej. Działanie docetakselu jest fazowo specyficzne. Lek oddziałuje na fazy S, M oraz dodatkowo na fazę G2 cyklu komórkowego. W artykule przedstawiono obecny stan wiedzy na temat molekularnych mechanizmów oddziaływania docetakselu na cykl komórkowy i dynamikę mikrotubul. Omówiono krótko także najnowsze dane dotyczące taksanów II i III generacji.
Słowa kluczowe:docetaksel • mikrotubule • tubulina • taksany II i III generacji
Summary
Docetaxel (Taxotere), a new semisynthetic taxoid, is a mitotic inhibitor, widely used in monotherapy or in combination with other anticancer drugs against many types of cancer. The structure and dynamics of microtubules as the main target for docetaxel activity inside the cell and thetaxane-binding site on β-tubulin are discussed. Microtubules are highly dynamic assemblies of α- and β-tubulin. They readily polymerize and depolymerize in cells and these dynamic behaviours are crucial to cell mitosis. Microtubule instability is attributed to their capability to hydrolyze GTP to GDP, which causes their depolymerization. Addition of new a-,b-tubulin heterodimer bound to GTP leads to tubulin polymerization, which increases the length of the microtubule. Docetaxel alters the polymerization dynamics of microtubules, which causes blockage of cell mitosis, and consequently induces apoptotic and non-apoptotic cell death. Docetaxel specifically acts on the S, M and G2 phases of the cell cycle. This paper reviews the current state of knowledge related to the molecular mechanisms of docetaxel action on the cell cycle and microtubule dynamics. In addition, a brief survey of the present state of research on the new generation (2nd and 3rd) of taxanes is presented.
Key words:docetaxel • microtubules • tubulin • 2nd and 3rd generation of taxanes
Wykaz skrótów:
AUC – pole pod krzywą stężenia leku (miara ilości leku jaka dociera do krążenia ogólnego w postaci niezmienionej) (area under the curve); CYP3A – izoenzym cytochromu P-450, rodzina 3, podrodzina A (cytochrome P450, family 3, subfamily A); CYP3A4 – izoenzym cytochromu P-450, rodzina 3, podrodzina A, izoforma 4 (cytochrome P450, family 3, subfamily A, isoform 4); DTX – docetaksel (docetaxel); MAPs – białka towarzyszące mikrotubulom (microtubule-associated proteins); MDR – oporność wielolekowa (multidrug resistance); MPF – czynnik promujący mitozę (maturation/mitosis promoting factor); MSAs – związki stabilizujące mikrotubule (microtubule-stabilizing agents); MTD – maksymalna tolerowana dawka (maximum tolerated dose); MTOC – centrum organizacji mikrotubul (microtubule – organizing centre); PTX – paklitaksel (paclitaxel); SAC – punkt kontrolny formowania wrzeciona kariokinetycznego (spindle assembly checkpoint); γ-TuRC – kompleks pierścieniowy gamma tubuliny (gamma-tubuline ring complex).
Wprowadzenie
Taksany (inaczej taksoidy) to jedne z najbardziej skutecznych leków przeciwnowotworowych nowej generacji. Należą do inhibitorów mitozy i stosowane są zarówno w monoterapii, jak i w skojarzeniu z innymi lekami przeciwnowotworowymi [1,41]. Głównym miejscem ataku taksanów są mikrotubule cytoszkieletu komórkowego. Interakcje pomiędzy taksanami i mikrotubulami zaburzają funkcje wrzeciona podziałowego, uniemożliwiają prawidłowe rozdzielenie chromatyd siostrzanych i chromosomów homologicznych podczas podziału komórki, co prowadzi do zahamowania mitozy. Działanie taksanów, tak jak innych inhibitorów mitozy, jest fazowo specyficzne i dotyczy faz S, G2 i M cyklu. Zatrzymanie cyklu komórkowego w G2/M uruchamia wiele mechanizmów, kierujących komórkę na drogę apoptozy [96]. Taksany mogą także wywoływać katastrofę mitotyczną komórek nowotworowych [52].
Taksany używane są w terapii neoadiuwantowej, mającej na celu remisję zmian pierwotnie nieoperacyjnych, oraz w terapii adiuwantowej, stosowanej po leczeniu chirurgicznym w celu wydłużenia czasu przeżycia. Obecnie chemioterapia przedoperacyjna jest postępowaniem standardowym w leczeniu miejscowo zaawansowanego raka piersi oraz raka zapalnego. Dotychczasowe badania eksperymentalne i kohortowe wskazują na wyraźne korzyści wynikające ze stosowania tego typu terapii m.in. ze względu na ograniczenie szybkości wzrostu guza pierwotnego po operacji oraz spadek ryzyka rozwoju oporności wielolekowej (multidrug resistance – MDR) w guzie pierwotnym [7,22,33]. Syntetyzowane są także liczne nowe pochodne taksanów, definiowane jako taksany II i III generacji. Znajdują się one jednak wciąż w fazie badań [56,84].
Stosowane klinicznie taksoidy paklitaksel (Taxol) i docetaksel (Taxotere) należą do taksanów I generacji. Mimo podobnej budowy oba leki znacznie różnią się aktywnością przeciwnowotworową i toksycznością. Dotychczasowe badania wykazały, że docetaksel (DTX), dzięki swoim unikalnym właściwościom i szerokim zakresie działania, przewyższa skutecznością paklitaksel (PTX). Większa aktywność przeciwnowotworowa docetakselu może wynikać z jego liniowej farmakokinetyki i dłuższej retencji w komórce [67]. W porównaniu z paklitakselem, docetaksel charakteryzuje się także większym powinowactwem do miejsc wiązania się z tubuliną, przez co jest bardziej cytotoksyczny. W warunkach in vitro również wykazano 1,3-12 razy większą cytotoksyczność DTX w różnych liniach komórek człowieka i myszy [66].
Budowa chemiczna taksanów
Różnice w budowie chemicznej docetakselu i paklitakselu wpływają w sposób istotny na ich właściwości przeciwnowotworowe (ryc. 1). Obecnie stosowane leki są półsyntetycznymi pochodnymi 10-deacetylobakatyny, związku uzyskiwanego z igieł cisu pospolitego (Taxus baccata) (pierwotnie paklitaksel otrzymywano z kory cisu krótkolistnego (Taxus brevifolia), co ze względu na rzadkie występowanie tego gatunku i jego wyjątkowo powolny wzrost było wysoce nieopłacalne) [10].
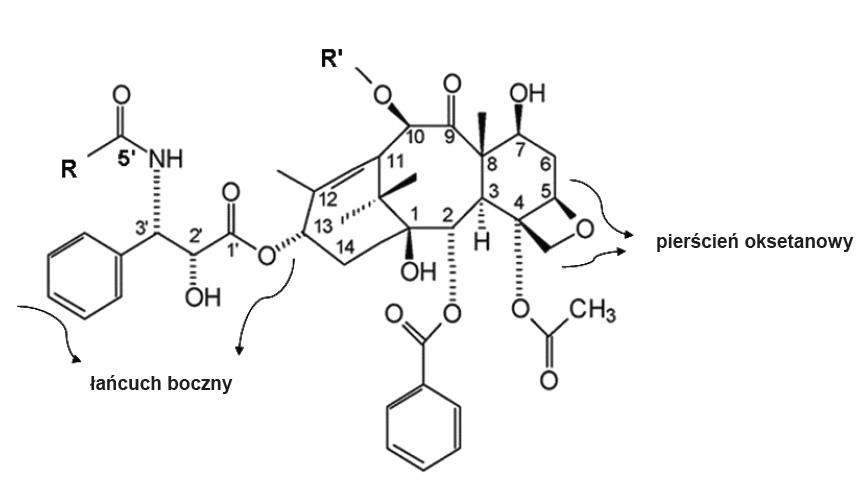
Ryc. 1. Struktura chemiczna taksanów
W cząsteczce docetakselu i paklitakselu można wyróżnić część rdzeniową, składającą się z 14-członowego pierścienia taksanowego oraz przyłączonego do niego w pozycji 4 i 5 pierścienia oksetanowego. Do części rdzeniowej w pozycji 13 pierścienia taksanowego przyłączony jest estrowo rozbudowany łańcuch boczny. Odgrywa on decydującą rolę w aktywności przeciwnowotworowej taksanów [69]. Pierścień taksanowy zawiera ponadto 2 podstawniki boczne: acetylowy w pozycji 4 i benzoilowy w pozycji 2. Odmienna budowa docetakselu i paklitakselu wynika z obecności różnych podstawników w pozycji 10 pierścienia taksanowego oraz w pozycji 5′ bocznego łańcucha. Charakterystyczne dla paklitakselu grupa acetylowa przy węglu 10 i podstawnik fenylowy przy węglu 5′ zostały w cząsteczce docetakselu zastąpione odpowiednio grupą hydroksylową i grupą alkoksylową (tabela 1). Docetaksel (N-benzoilo-N-tert-butoksykarbonylo-10-deacetylo-taksol) ma masę cząsteczkową 807,9, jest lipofilny i nierozpuszczalny w wodzie [13].
Tabela 1. Podstawowe różnice w budowie chemicznej docetakselu i paklitakselu

Schematy podawania, farmakokinetyka i metabolizm docetakselu
Stosowane w monoterapii dawki docetakselu zawarte są w przedziale 35 mg/m2-100 mg/m2, w zależności od sposobu podawania. Lek najczęściej aplikowany jest w jednogodzinnym wlewie dożylnym, co 21 dni. Liczba cykli zależy od stanu pacjenta i stopnia zaawansowania choroby [35,72]. Istnieją również tygodniowe schematy podawania, wtedy dawka DTX zredukowana jest do 35-40 mg/m2. DTX stosowany jest co tydzień przez 6 kolejnych tygodni, po czym następuje 2-tygodniowa przerwa lub lek podawany jest co tydzień przez kolejne 3 tygodnie, po czym następuje tygodniowa przerwa [18,26,82]. Przy dawce zredukowanej czas infuzji może wynosić 30 min lub 1 h [2,45]. W praktyce szuka się takiego schematu podawania leku, który cechuje się dużą skutecznością oraz najmniejszym ryzykiem pojawienia się działań niepożądanych [38].
Maksymalna tolerowana dawka (MTD) docetakselu wynosi 100 mg/m2. Po krótkim dożylnym podaniu dawki MTD stwierdza się liniową farmakokinetykę leku, według modelu trójkompartmentowego, o następujących okresach połowicznego półtrwania: t1/2α 4-5 min, t1/2β 36-60 min oraz t 11,1 godz. Docetaksel charakteryzuje się dość dużą objętością dystrybucji, wynoszącą 113 L. Pole pod krzywą stężenia leku (area under the curve – AUC) jest wprost proporcjonalne do podanej dawki i nie zależy od klirensu docetakselu [8]. Stwierdzono, że przy podaniu DTX w schemacie tygodniowym (po jednym cyklu chemioterapii) w dawce 35 mg/m2, stężenie leku w surowicy krwi, bezpośrednio po podaniu, wynosiło 1 nM i utrzymywało się przez 7 dni. Po podaniu DTX w dawce 75 mg/m2 według schematu trzytygodniowego, stężenie leku w surowicy utrzymywało się na poziomie 0,5 nM przez 21 dni [6].
Przeprowadzone dotychczas badania wykazują, iż zarówno u ludzi, jak i u szczurów powstają takie same metabolity docetakselu, co może sugerować, że metabolizm leku przebiega w taki sam, albo bardzo podobny sposób [59,78]. Metabolizm docetakselu zachodzi głównie w wątrobie z udziałem monooksygenaz z rodziny cytochromu P-450. Największą rolę w tym procesie odgrywa podrodzina CYP3A cytochromu P-450 i należące do niej izoformy CYP3A4 oraz CYP3A5 [83]. W wyniku działania tego enzymu powstają 4 metabolity o znacznie mniejszej cytotoksyczności niż cząsteczka macierzysta [50]. W pierwszym etapie metabolizmu jedna z grup metylowych reszty tert-butylowej łańcucha bocznego w pozycji 13 ulega hydroksylacji do reszty alkoholowej. Hydroksypochodna ulega cyklizacji poprzez niestabilny aldehyd do dwóch diastereoizomerów hydroksyoksazolidynonowych lub może też ulec utlenieniu i cyklizacji do oksazolidynodionu (ryc. 2). Nie zidentyfikowano natomiast metabolitów związanych z hydroksylacją 14-członowego pierścienia rdzenia cząsteczki. Powstałe metabolity wydalane są z organizmu, głównie z żółcią, i w znikomym stopniu z moczem. Dotychczas nie udowodniono eksperymentalnie możliwości sprzęgania metabolitów docetakselu z kwasem glukuronowym ani z jakąkolwiek inną cząsteczką [14,70].
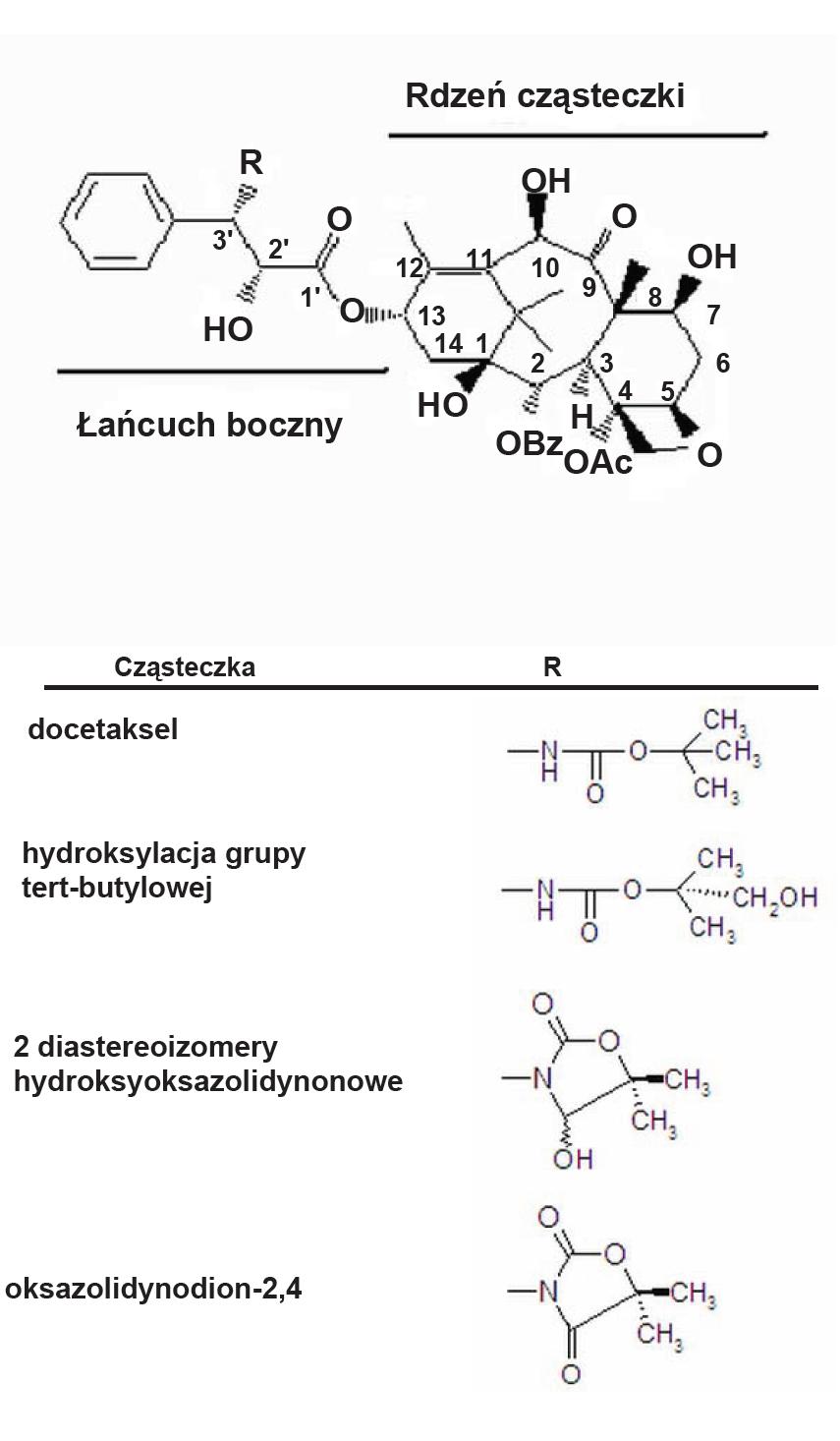
Ryc. 2. Metabolizm docetakselu (wg [78] -zmienione)
Działanie docetakselu na mikrotubule
Budowa mikrotubul
Docetaksel w komórce działa głównie na mikrotubule, dynamiczne rurkowate struktury zbudowane przeważnie z 13 równoległych protofilamentów. Liczba protofilamentów może się wahać od 9 do 16 [12]. Mają one kształt cylindra o średnicy około 25 nm i osiągają długość do kilkunastu mikrometrów. Protofilamenty powstają w wyniku polimeryzacji globularnych białek α-tubuliny (m.cz. 55 000-57 000 Da) i β-tubuliny (m.cz. 53 000 Da). Białka te łączą się ze sobą wiązaniem niekowalencyjnym tworząc heterodimer wielkości 3,5×4 nm. Jest on podstawową jednostką strukturalną protofilamentu. Heterodimery w protofilamencie układają się liniowo i naprzemiennie w ten sposób, że na jednym z jego końców znajduje się α-tubulina, a na drugim β-tubulina [4,53]. Zróżnicowana struktura chemiczna końców protofilamentów nadaje mikrotubulom strukturalną biegunowość, która jest niezwykle istotna zarówno dla montażu i demontażu mikrotubul, jak i dla pełnionych przez nie funkcji, zwłaszcza w określaniu kierunku transportu wewnątrzkomórkowego, związanego ze strukturami błonowymi komórki.
W komórkach zwierzęcych mikrotubule formowane są w centrosomach, które pełnią rolę centrum organizacji mikrotubul (microtubule-organizing centre – MTOC) i regulatora progresji cyklu komórkowego. Za zakotwiczenie mikrotubul i inicjację nukleacji (polimeryzację heterodimeru α-, β-tubuliny) odpowiedzialnych jest kilka białek (γ-tubuliny, pericentryna i nineina), umiejscowionych w pericentriolarnym materiale centrosomu, w pobliżu centriol. Tworzą one strukturę pierścieniową, tzw. pierścień nukleacyjny zbudowany γ-tubuliny (kompleks γ-TuRC) (gamma-tubuline ring complex), z którego „wyrastają” nowo powstające mikrotubule [20,89]. Kompleks γ-TuRC swoją strukturą chemiczną naśladuje budowę końca (+) mikrotubul, umożliwiając tym samym przyłączenie się końca (-) heterodimeru α-, β-tubuliny do pierścienia nukleacyjnego i dalszą polimeryzację przez dodawanie kolejnych heterodimerów [25,51]. Rosnąca mikrotubula przyłączona jest do pierścienia nukleacyjnego końcem (-), a koniec (+), znajdujący się w cytoplazmie, skierowany jest w stronę błony komórkowej (ryc. 3). Zakotwiczenie mikrotubuli w MTOC działa stabilizująco, gdyż umożliwia jej szybkie przyrastanie od strony końca (+) [61]. Mikrotubule nie zawsze są tworzone w połączeniu z kompleksem γ-TuRC centrosomu (np. aksony, dendryty). W pewnych warunkach centra nukleacji mogą powstawać spontanicznie w różnych regionach komórki [94].
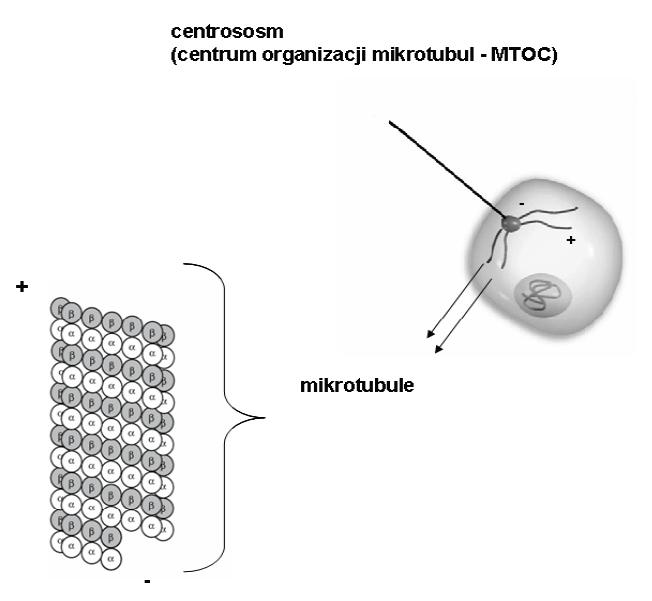
Ryc. 3. Budowa i umiejscowienie mikrotubul w komórce (schemat)
W komórce mikrotubule tworzą cztery rodzaje struktur: sieć interfazową, wrzeciono podziałowe, aksony rzęsek i wici oraz centriole. W cytoplazmie komórki interfazowej mikrotubule występują w postaci pojedynczych lub rozproszonych wiązek, ułożonych wzdłuż długiej osi komórki.
Oprócz α, β i γ-tubuliny zidentyfikowano jeszcze inne rodzaje tubuliny, są to: δ-, ε-, ζ- i η-tubulina, które biorą udział w tworzeniu wrzeciona podziałowego. Nie występują one jednak we wszystkich organizmach eukariotycznych, np. u człowieka nie znaleziono ζ- i η-tubuliny. Znane są różne izoformy wymienionych rodzajów tubuliny. W komórkach człowieka są to: TUBA1A, TBA1B, TUBA1C, TUBA3C, TUBA3D, TUBA3E, TUBA4A i TUBA8 a-tubuliny; TUBB, TUBB1, TUBB2A, TUBB2B, TUBB2C, TUBB3, TUBB4, TUBB4Q i TUBB6 β-tubuliny; TUBG1, TUBG2, TUBGCP2, TUBGCP3, TUBGCP4, TUBGCP5, TUBGCP6 γ-tubuliny oraz TUBD1 δ-tubuliny i TUBE1 ε-tubuliny [16]. Wyróżnia się ponadto 7 izotypów β-tubuliny, które wykazują różną ekspresję, funkcję i strukturę (głównie C-końcowej sekwencji aminokwasów) [44].
Homologiem tubuliny (10-18% homologiczność na poziomie aminokwasów) w komórkach prokariotycznych jest białko FtsZ. Występuje ono również w plastydach i mitochondriach niektórych organizmów eukariotycznych. Pod względem struktury trzeciorzędowej FtsZ wykazuje duże podobieństwo do tubuliny, a jego polimeryzacja, podobnie jak polimeryzacja tubuliny, jest zależna od GTP [42].
Funkcje mikrotubul w komórce
Mikrotubule wraz z mikrofilamentami i filamentami pośrednimi tworzą cytoszkielet komórki eukariotycznej. Pełnią w niej m.in. rolę masztów rozporowych, gdyż są jedynym elementem cytoszkieletu zdolnym do przenoszenia naprężeń ściskających. Stanowią także szlaki, wzdłuż których odbywa się transport wewnątrzkomórkowy i przemieszczają się dyneiny, kinezyny i miozyny – enzymatyczne białka motoryczne (białka kroczące, transportujące, motory molekularne), wykazujące zdolność do generowania ruchu za pomocą wykorzystania energii chemicznej pochodzącej z hydrolizy ATP [91]. Mikrotubule, wraz z towarzyszącymi im białkami motorycznymi, odgrywają istotną rolę w rozmieszczeniu i utrzymywaniu stałego położenia niektórych organelli komórkowych, np. mitochondriów, plastydów, cystern w diktiosomie itp., a także w procesach związanych ze zmianą kształtu komórki, takich jak ruch, podział, różnicowanie, transformacja nowotworowa czy apoptoza [36]. Mikrotubule biorą także udział w kaskadzie reakcji sygnalizacyjnych w komórce. Stwierdzono, że odpowiedź komórki na sygnały zewnątrzkomórkowe, przekazywane za pośrednictwem receptorów znajdujących się na powierzchni komórki, odbywa się m.in. z udziałem cząsteczek zaliczanych do rodziny białek Rho, przejawiających aktywność GTP-azową. Szlak sygnałowy z wykorzystaniem tych białek wpływa na dynamikę mikrotubul, która z kolei jest ważnym sygnałem w procesach podziału komórki, jej polaryzacji czy ruchu [30].
Budowa heterodimeru α-, β-tubuliny oraz miejsca przyłączania się docetakselu
Każdy z monomerów heterodimeru α-, β-tubuliny ma trzy główne domeny funkcjonalne: N-końcową, która uczestniczy w procesie łączenia się mikrotubul z nukleotydami (GTP lub GDP), centralną, zaangażowaną w naprzemienne ułożenie heterodimerów α-, β-tubuliny, tworzących łańcuch zwany protofilamentem oraz C-końcową, niezbędną w procesie przyłączania się związanych z mikrotubulami białek MAPs (microtubule-associated proteins) (ryc. 4). Białka MAPs występują na powierzchni mikrotubul i uczestniczą w procesie ich łączenia się między sobą [58].

Ryc. 4. Struktura krystalograficzna dimeru tubuliny z zaznaczeniem miejsc wiązania się z nukleotydami (miejsce E oraz N) i z taksanami – opis w tekście (wg [15] – zmieniono)
Domena N-końcowa zawiera reszty 1-205 i tworzy strukturę zwaną motywem Rossmanna (Rossman fold). Wraz z sześcioma równoległymi harmonijkami β (B1-B6) oraz helisami α (H1-H6) bierze ona udział w procesie łączenia się mikrotubuli z GTP lub GDP. Domenę centralną budują reszty 206-381, tworzące cztery różne harmonijki β (B7-B10) oraz cztery helisy α (H7-H10). Istotnym elementem domeny centralnej jest pętla, zawierająca aminokwasy 272-288, zwana pętlą M, łącząca harmonijkę B7 z helisą H9. Pętla M odrywa ważną rolę w stabilizacji heterodimeru oraz całego protofilamentu. Domena C-końcowa rozpoczyna się od reszty 382, kończy aminokwasem 440 i składa się z dwóch przeciwrównoległych helis (H11 i H12) [15,37,58]. Reszta 385 odgrywa ważną funkcję w procesie przyłączania się białek MAPs, takich jak tau, Map2, statmina, CENP-E (kinezyna 7) i kinezyna EgP [48] (ryc. 4).
W strukturze heterodimeru α-, β-tubuliny występują ponadto dodatkowe, niewielkich rozmiarów α helisy. W tubulinie-α są to helisy H3′, H11′, natomiast w tubulinie-β H1′, H2′, H2”, H3′, H9′ i H11′ [43].
Docetaksel wiąże się z domeną centralną tubuliny-β, w obszarze obejmującym hydrofobową szczelinę umiejscowioną blisko powierzchni białka [15,63,77]. Między tubuliną a taksanem występuje wiele oddziaływań niekowalencyjnych, do których zaliczyć można oddziaływania hydrofobowe, siły Van der Waalsa oraz wiązania wodorowe (ryc. 5).
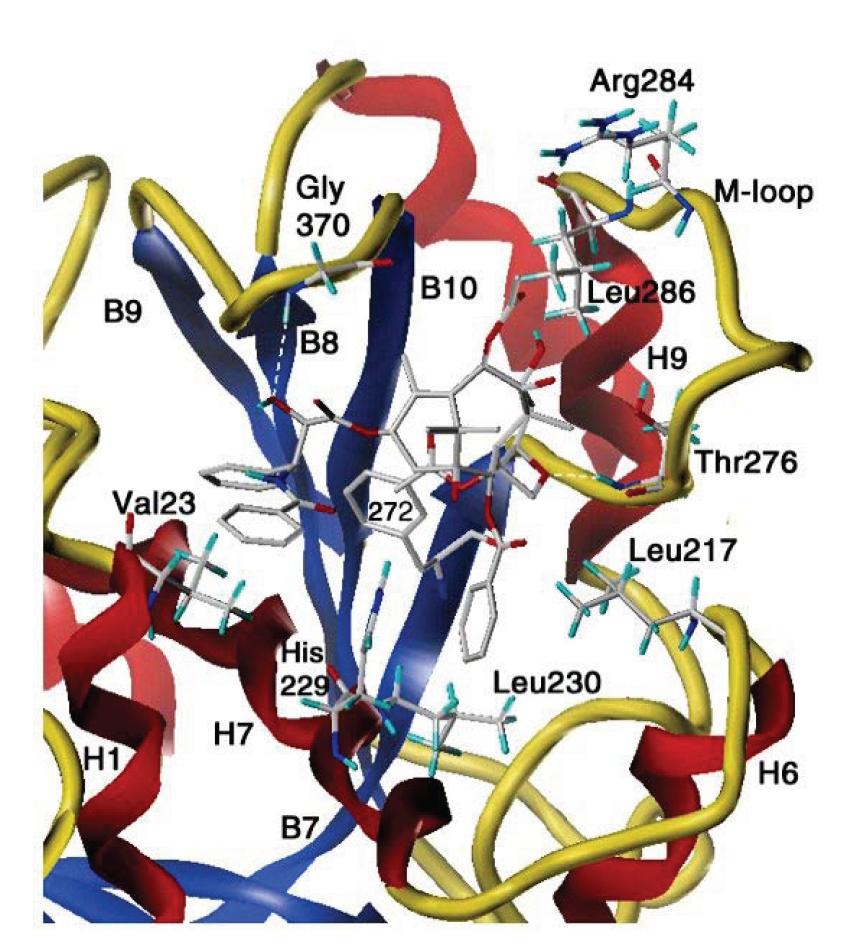
Ryc. 5. Szczegółowa budowa miejsca wiązania taksanów z β-tubuliną – opis w tekście (wg [77] za zgodą autorów)
Model przestrzennego dopasowania taksanów do tubuliny-b, zaproponowany przez Rao i wsp. [63], zakłada, że podstawnik w pozycji 3′ taksanów po przyłączeniu taksanu do tubuliny-β znajduje się w bliskim kontakcie z aminokwasami 1-31, natomiast ugrupowanie benzoilowe, występujące w pozycji 2, umiejscowione jest w pobliżu aminokwasów 217-233. Dodatkowo grupa hydroksylowa w pozycji 7 pierścienia taksanowego tworzy wiązanie z 282 argininą [63].
W 2001 r. opisano miejsce „dokowania się” paklitakselu do hydrofobowej szczeliny występującej w tubulinie-β. W procesie tym zaangażowane są: helisa H1, pętla łącząca helisy H6 i H7, helisa H7, pętla M oraz pętla łącząca struktury β: B9 i B10. Walina 23 oraz asparagina 26, wchodzące w skład aminokwasów budujących helisę H1, tworzą hydrofobowe oddziaływanie z podstawnikiem w pozycji 3′ łańcucha bocznego cząsteczki leku. Podobnie alanina 233 oraz seryna 236, występujące w helisie H7 znajdują się w bliskim kontakcie z tym podstawnikiem. Grupa benzoilowa w pozycji 2 pierścienia taksanowego znajduje się w hydrofobowym środowisku utworzonym przez następujące aminokwasy: leucynę 217 i 219, w pętli łączącej helisę H6 z H7 oraz histydynę 229 i leucynę 230, znajdujące się w helisie H7. Dodatkowe oddziaływanie utworzone jest przez fenyloalaninę 272 harmonijki β: B7 [77].
Stwierdzono, że mutacja powodująca zamianę tego aminokwasu w walinę skutkuje pojawieniem się częściowej oporności na paklitaksel in vitro [21]. Pierścień oksetanowy taksanu wpływa na aminokwasy: prolinę 274, leucynę 275, treoninę 373, serynę 277 i argininę 278 pętli M. Szczelina, w której umiejscawia się taksan ograniczona jest przez następujące aminokwasy pętli łączącej harmonijki b: B9 z B10: prolina 360, arginina 369, glicyna 370 i leucyna 371 (ryc. 5) [43,77].
Przedstawiony w 2006 r. model przestrzennego ułożenia taksanów w hydrofobowej kieszonce zakłada, że leki z tej grupy po przyłączeniu się do β-tubuliny przyjmują formę litery T zwaną również „formą motyla” (butterfly model) [3]. Model ten tłumaczy oddziaływania występujące między taksanami a tubuliną, zakładając, że biorą w nich udział zarówno pierścień taksanowy, jak i łańcuch boczny leku. Stwierdzono, że ugrupowania znajdujące się w pozycji 3′ i w pozycji 2 cząsteczki taksanu oddziałują hydrofobowo z fragmentami helis H1, H6 i H7 oraz z pętlą występującą między helisami H6 i H7. Dodatkowe oddziaływanie pojawia się między podstawnikiem w pozycji 3′ a strukturami B8 i B10. Istotna dla aktywności taksanów grupa hydroksylowa, znajdująca się w pozycji 2′ łańcucha bocznego tworzy wiązanie wodorowe z grupą NH pętli łączącej struktury B9 i B10. Dodatkowo grupa metylowa w pozycji 12 głównego pierścienia taksanów lokuje się blisko leucyny 371 umiejscowionej w tej pętli. Grupa metylowa w pozycji 8 pierścienia taksanowego skierowana jest w stronę pętli M i oddziałuje siłami Van der Waalsa z dwoma aminokwasami, tj. treoniną 276 i glicyną 281, które znajdują się blisko końca C β-tubuliny. Grupa acetylowa w pozycji 4 pierścienia taksanowego znajduje się bezpośrednio nad szczeliną utworzoną przez 10 aminokwasów, z których część występuje w pętli M [3,53,77]. Ostatnie badania potwierdzają, że taksan, po przyłączeniu się do tubuliny, przyjmuje kształt litery T [93].
Dynamiczna niestabilność mikrotubul a mechanizm działania docetakselu
Mikrotubule w komórce znajdują się w stanie dynamicznej niestabilności. Ulegają ciągłemu wydłużeniu (polimeryzacji) lub skracaniu (depolimeryzacji), mogą też całkowicie się rozpaść lub zacząć formować od nowa (ryc. 6). Dynamiczna niestabilność mikrotubul wynika ze zdolności tubuliny do przyłączania i hydrolizowania guanozynotrifosforanu (GTP) i ma istotne znaczenie dla prawidłowego funkcjonowania komórki. Stwierdzono, że przyłączenie się docetakselu do mikrotubul zaburza ich organizację i powoduje zachwianie równowagi dynamicznej niestabilności na skutek przewagi procesów polimeryzacji nad depolimeryzacją. Dochodzi do zaburzenia wielu procesów, w których mikrotubule odgrywają główną rolę, takich jak podział komórki czy transport substancji [40]. Ze względu na sposób działania docetaksel zaliczany jest zatem do związków stabilizujących mikrotubule MSAs (microtubule-stabilizing agents) [96].
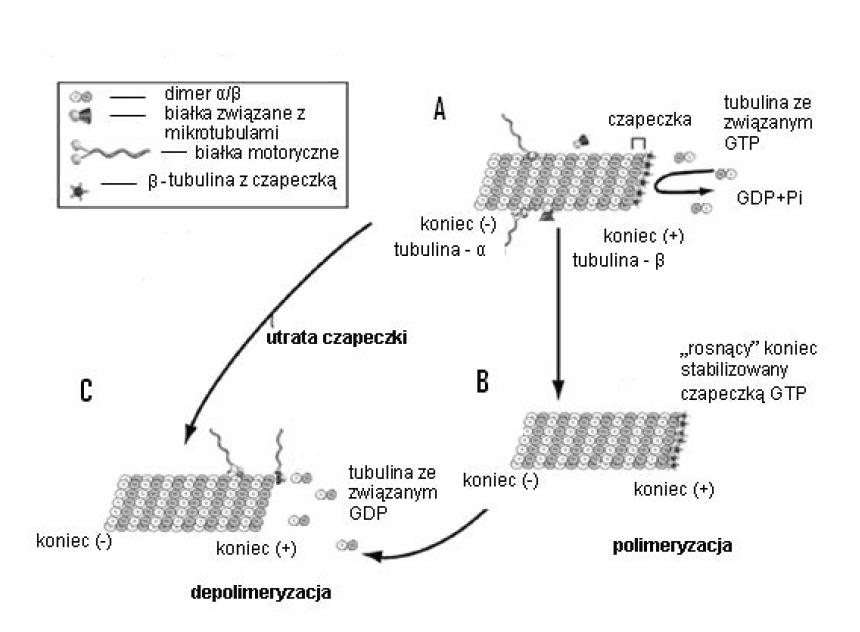
Ryc. 6. Procesy polimeryzacji i depolimeryzacji mikrotubul; A – w miejscu E β-tubuliny przyłącza się GTP, B – hydroliza GTP następuje powoli, nowe heterodimery ze związanym GTP mogą się przyłączyć do końca (+), powstaje ochronna „czapeczka”, C – hydroliza GTP zachodzi bardzo szybko, następuje utrata „czapeczki”, a dimery z przyłączonym GDP „odrywają się” od mikrotubuli (wg [33] – zmienione)
W heterodimerze istnieją dwa miejsca wiążące GTP: miejsce zdolne do hydrolizy GTP, tzw. miejsce E (exchangeable site) w β-tubulinie i niemające takiej zdolności miejsce N (non-exchangeable site) w α-tubulinie [47]. Główne źródło energii w procesie polimeryzacji mikrotubul stanowi hydroliza GTP. Wolne heterodimery α-, β-tubuliny łączą się z cząsteczką GTP i w tej postaci dołączane są do rosnącej (polimeryzowanej) mikrotubuli. Procesy zachodzące w obrębie miejsca E mogą mieć dwojaki charakter. W zależności od warunków (np. dostępność GTP i wolnej tubuliny) hydroliza GTP może zachodzić przed lub po dołączeniu następnego heterodimeru do rosnącej mikrotubuli. Proces hydrolizy przebiega wolno, gdy nukleotyd połączony jest z niezwiązanym z mikrotubulą (wolnym) heterodimerem α-, β-tubuliny lub gdy mikrotubula rośnie powoli. Wtedy związana z heterodimerem α-, β-tubuliny cząsteczka GTP może ulec hydrolizie do GDP zanim przyłączy się kolejny heterodimer. Odłączenie się od protofilamentów końcowych podjednostek, związanych z GDP, może spowodować depolimeryzację i skrócenie mikrotubuli.
W warunkach nadmiaru wolnego GTP i wolnej tubuliny proces dołączania kolejnych podjednostek α-, β-tubuliny-GTP do końca (+) protofilamentów zachodzi szybko i związane GTP nie zdąży ulec hydrolizie. Przyłączone podjednostki zakrywane są więc sukcesywnie przez kolejne GTP-heterodimery. W wyniku tego powstaje fragment protofilamentu zawierający przyłączony GTP. Fragment ten pełni rolę ochronnej „czapeczki” (GTP-cap), która blokuje odłączania się wewnętrznych heterodimerów związanych z GDP i tym samym promuje polimeryzację i wydłużanie się mikrotubul. Reszta protofilamentów, od strony końca minus, zawiera heterodimery połączone z GDP [34,87].
Konformacja heterodimeru α-, β-tubuliny zależy od tego czy β-tubulina połączona jest z dinukleotydem czy z trinukleotydem. Po związaniu się z GTP przybiera konformację prostą (niezakrzywioną). Gdy ulegnie hydrolizie do GDP przyjmuje konformację zakrzywioną, tzw. formę D. Heterodimery ze związanym GDP są bardzo niestabilne i odłączają się od mikrotubuli. W wyniku tego ulega ona depolimeryzacji i skróceniu. Utrata kolejnych heterodimerów może przebiegać do całkowitego zaniku filamentu. Mikrotubula może też ulec tylko częściowej depolimeryzacji i ponownie zacząć się wydłużać [49]. Procesy polimeryzacji i depolimeryzacji mikrotubul i ich pozorne nieuporządkowanie znajdują się pod stałą kontrolą wielu białek. Grupa białek towarzyszących mikrotubulom – MAPs bierze udział w stabilizacji struktury mikrotubul poprzez łączenie polimerów i ułatwianie oddziaływań z innymi składnikami komórki. Białko Map-4 przyłączając się do mikrotubul inicjuje proces ich polimeryzacji i przyspiesza proces nukleacji, czyli formowania się nowych kompleksów w miejscu MTOC, a jednocześnie hamuje odłączanie się tubuliny z końców miktotubul. Fosforylacja białek MAPs w wyniku obróbki potranslacyjnej prowadzi do ich odłączania się od miktotubul, co wywołuje niestabilność tych struktur [31]. Do MAPs należą też niskocząsteczkowe białka tau, które pośredniczą zarówno w wiązaniu się mikrotubul między sobą, jak i z innymi elementami cytoszkieletu [86]. Ważną rolę w utrzymaniu funkcji miktotubul w komórce odgrywają dwie grupy białek, dyneiny i kinezyny, wykazujące aktywność ATP-az. Dzięki energii uzyskanej z hydrolizy ATP białka te zmieniają swoją konformację i mogą się przesuwać wzdłuż mikrotubul. Zmiana umiejscowienia dyneiny na mikrotubuli ma znaczenie w procesach ruchu migawek i rzęsek oraz w procesach transportu związków chemicznych w komórce [80].
Protofilamenty formujące mikrotubule są proste i układają się równolegle. Połączone z GDP heterodimery α-, β-tubuliny, które przybierają konformację zakrzywioną (postać D) utrzymywane są w konformacji wyprostowanej w protofilamentach na skutek oddziaływań z heterodimerami sąsiednich protofilamentów.
Wyróżnia się dwa rodzaje oddziaływań: poprzeczne i podłużne. Stwierdzono, że mechanizm działania taksanów na mikrotubule polega m.in. na wzmocnieniu istniejących oddziaływań, zarówno poprzecznych jak i podłużnych między heterodimerami α-, β-tubuliny [5,17].
Poprzeczne oddziaływania pomiędzy heterodimerami odbywają się z udziałem pętli M jednego heterodimeru i pętli łączącej helisę H1 z harmonijką B2 (H1-B2). Ważne dla utrzymania oddziaływań poprzecznych są również helisy H3 i H10 oraz pętla łącząca struktury: H2-B2 (ryc. 7). Połączenia podłużne między heterodimerami budującymi protofilament utrzymywane są za pomocą oddziaływań między helisą H10 i helisą H3 [32,39].

Ryc. 7. Miejsce połączenia między sąsiadującymi heterodimerami – opis w tekście (wg [15] – zmienione)
Stwierdzono, że DTX wpływa stabilizująco na mikrotubule przez oddziaływanie na pętlę M tubuliny-β. Pętla ta może ulegać różnym zmianom konformacyjnym, w zależności od stanu uporządkowania mikrotubul. Części górna i dolna pętli M działają jak zawiasy, umożliwiające zmianę kąta połączenia sąsiadujących heterodimerów. Zmiana struktury przestrzennej pętli M pociąga za sobą zmianę tego kąta. Stwierdzono, że pętla M w α-tubulinie stabilizowana jest przez długą pętlę aminokwasową, łączącą strukturę B9 z B10. Pętla M w β-tubulinie nie jest stabilizowana, a jej ruchomość ma istotny wpływ na utrzymanie stanu dynamicznej niestabilności mikrotubul. Zmniejszenie ruchomości skutkuje zahamowaniem depolimeryzacji mikrotubul [43]. Docetaksel stabilizuje konformację pętli M, odgrywa więc w β-tubulinie rolę pętli łączącej strukturę B9 z B10 [77]. Badania z wykorzystaniem komputerowego modelowania ruchomości poszczególnych składowych heterodimeru ujawniły, że po przyłączeniu taksanu do β-tubuliny nastąpił drastyczny spadek sprężystości struktur znajdujących się w otoczeniu leku. Zaobserwowano głównie usztywnienie w okolicy aminokwasów 214-224 oraz 272-285 (pętla M). Zauważono również ograniczenie możliwości zmian konformacyjnych m.in. w rejonie zawierającym aminokwasy 35-44, wchodzące w skład pętli łączącej helisę H1 ze strukturą B2 [37].
Zaburzenie procesów polimeryzacji i depolimeryzacji, spowodowane przez docetaksel, wynikać może również z zahamowania zmian strukturalnych helisy H3. Konformacja helisy H3 jest wrażliwa na hydrolizę GTP. Gdy w miejscu E występuje GTP połączenie z sąsiadującym heterodimerem – zachodzące z udziałem helisy H3 – jest mocne, a wraz z hydrolizą GTP następuje osłabienie oddziaływań i może dojść do odrywania się heterodimeru od mikrotubuli. Docetaksel stabilizując konformację helisy H3 sprawia, że staje się ona niepodatna na zmiany spowodowane hydrolizą GTP. Jednak działanie taksanu stabilizujące mikrotubule może wynikać z usztywnienia określonej konformacji pętli H1-S2 promującej połączenia poprzeczne między heterodimerami [37,92].
Znaczenie mikrotubul w cyklu komórkowym i jego zaburzenia pod wpływem docetakselu
Mikrotubule odgrywają szczególną rolę w procesie podziału komórki i segregacji chromosomów do komórek potomnych [49]. Wyróżnia się trzy typy mikrotubul: kinetochorowe, których funkcja polega na łączeniu się końcem (+) z kinetochorami chromatyd siostrzanych, międzybiegunowe, których końce wystają z przeciwnych biegunów komórki oraz gwiaździste (astralne), biorące udział w zakotwiczeniu wrzeciona podziałowego we właściwym miejscu w komórce. Łączą one centrosomy z częścią podbłonową, tym samym biorą udział w ich stabilizacji. W chwili wejścia komórki w fazę podziału interfazowe mikrotubule, które stanowiły stabilny element cytoszkieletu, biorący udział w transporcie związków w komórce, ulegają depolimeryzacji. W ich miejsce pojawiają się nowe mikrotubule, znacznie bardziej dynamiczne, tworzące włókna wrzeciona podziałowego [24].
W komórkach eukariotycznych proces mitozy rozpoczyna się wraz z pojawieniem się czynnika promującego fazę mitozy – MPF (maturation/mitosis promoting factor) utworzonego z kompleksów między podjednostką regulatorową cykliny B1 i zależną do cyklin kinazą (cdk1) p34cdc2 [54]. We wczesnej profazie chromosomy ulegają kondensacji i wyodrębniają się chromatydy siostrzane. Białka aparatu podziałowego np. kinezyna Eg5 (KSP) uczestniczą w procesie rozdzielania się chromosomów i formowania dwubiegunowych włókien podziałowych [74].
U większości komórek zwierzęcych każde włókno podziałowe jest tworzone w obrębie centrosomu, który ułatwia właściwą organizację przestrzenną mikrotubul. Docetaksel powoduje zaburzenia organizacji centrosomu. Stwierdzono, że lek ten był najbardziej cytotoksyczny dla nowotworowych komórek linii HeLa w fazie S cyklu komórkowego. W komórkach tych docetaksel spowodował odłączenie się od centrioli otaczającej je macierzy, co skutkowało pojawieniem się w fazie G2 wrzeciona o zaburzonej strukturze [60].
Klasyczna hipoteza tłumacząca tworzenie wrzecion podziałowych (tzw. koncepcja search-and-capture) zakłada, że mikrotubule wyrastające promieniście z centrosomów, nieustannie eksplorują przestrzeń komórki w trakcie kolejnych cykli polimeryzacji i depolimeryzacji. W przypadku kontaktu końca (+) mikrotubuli z kinetochorem dochodzi do stabilizacji mikrotubuli na skutek zahamowania jej depolimeryzacji, co powoduje powstanie stosunkowo trwałego połączenia chromosomu z biegunem komórki. Z czasem coraz więcej mikrotubul tworzy podobne połączenie wykorzystując pulę wolnej tubuliny w komórce, jednocześnie maleje liczba mikrotubul astralnych. W efekcie formuje się typowe wrzeciono: chromosomy połączone są wiązkami mikrotubul (chromosomalnych i kinetochorowych) z oboma biegunami komórki [64].
Wykazano, że w komórkach traktowanych docetakselem powstaje wrzeciono kariokinetyczne o zmienionej strukturze, co wpływa na funkcjonowanie aparatu mitotycznego. Nieprawidłowe tworzenie się wrzeciona kariokinetycznego może wynikać z zaburzeń w organizacji centrosomu wywołanych przez docetaksel. Na jednym biegunie komórki tworzą się dwa centrosomy, podczas gdy drugi biegun pozostaje bez centrosomu. W wyniku tego następuje nieprawidłowy rozdział chromosomów do komórek potomnych. Efektem końcowym jest utworzenie aneuploidalnych komórek potomnych [71]. Docetaksel może również wpływać na białka, których funkcją jest stabilizacja oddziaływań między centrosomami a mikrotubulami. Wykazano, że istotnym białkiem, biorącym udział w tej stabilizacji jest białko NuMa (nuclear mitotic apparatus). Paoletti i wsp. [60] wykazali, że w komórkach linii HeLa, traktowanych DTX, białko to preferencyjnie umiejscawia się na biegunie pozbawionym centrosomów. Nierównomierny rozkład NuMa z przewagą cząsteczek na biegunie pozbawionym centrosomów w wyniku działaniu DTX wykazały również badania Sakaushi i wsp. [71]. Usytuowanie białka NuMa na jednym z biegunów komórki ma znaczenie w jej asymetrycznym podziale [62].
Poprawność połączeń między kinetochorami i mikrotubulami tworzącymi wrzeciono jest nadzorowana przez kompleks białek kontrolujących powstawanie wrzeciona kariokinetycznego SAC (spindle assembly checkpoint). W chwili pojawienia się stabilnego połączenia, komórka wkracza w anafazę, w której następuje podział chromosomów [65]. Funkcja punktu kontrolnego SAC jest upośledzona w komórkach nowotworowych, dlatego też mogą przezwyciężyć zatrzymanie cyklu na tym etapie [52,90]. W rezultacie komórka opuszcza fazę mitozy z opóźnieniem w tzw. poślizgu mitotycznym (mitotic slippage), wkracza w fazę pseudo-G1 bez cytokinezy i zostaje zatrzymana w punkcie kontrolnym (restrykcyjnym) G1/S. Tym samym nie może wejść w fazę S, co zapobiega procesowi poliploidyzacji. Poślizg mitotyczny indukuje proces apoptozy [79].
Docetaksel aktywuje punkt kontrolny SAC. Jest to spowodowane nieprawidłową strukturą i dynamiką mikrotubul, wywołaną przyłączeniem się leku do β-tubuliny. W zależności od stężenia leku w komórce obserwuje się dwa mechanizmy działania, wynikające z aktywacji punktu SAC (ryc. 8). Małe stężenia leku (2-4 nM) powodują krótkotrwale zatrzymanie komórki w punkcie SAC i anomalie w podziale mitotycznym, prowadzące do aneuploidalności komórki. Przy większych stężeniach leku (100 nM) następuje natomiast dłuższe zatrzymanie komórki w punkcie kontrolnym SAC, skutkujące poślizgiem mitotycznym i opóźnionym wyjściem z mitozy. Powoduje to przejście komórek tetraploidalnych w fazę pseudo-G1 i uruchomienie szlaku apoptozy [29].
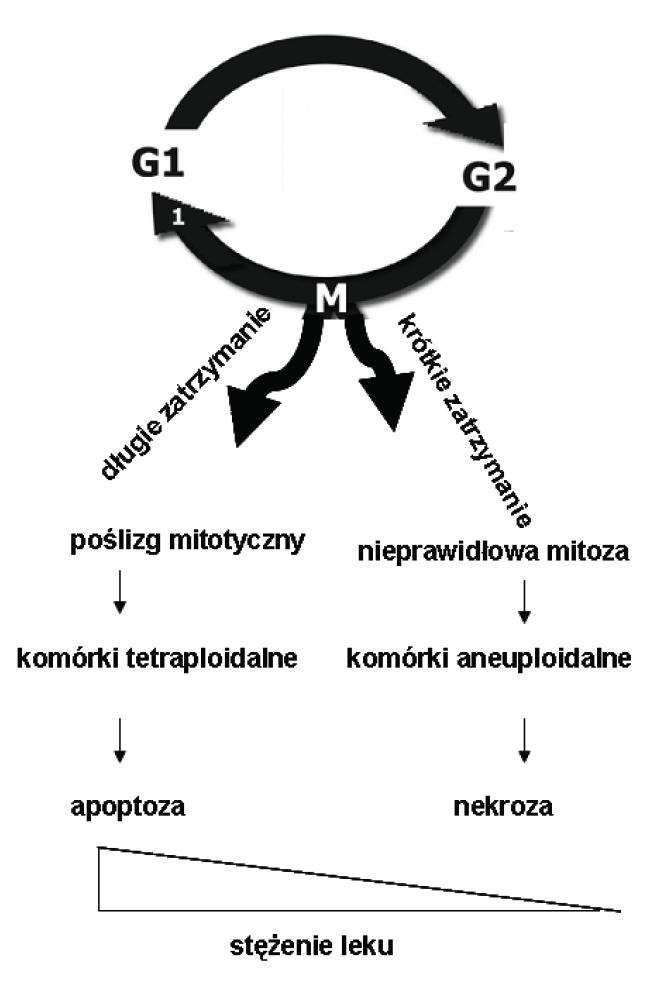
Ryc. 8. Schemat działania docetakselu na komórki, w zależności od stężenia – opis w tekście
Badania z zastosowaniem 100 nM stężenia DTX w komórkach linii MCF-7 wykazały, że w pierwszych godzinach hodowli następował wzrost ekspresji genów kodujących białka uczestniczące w regulacji funkcji punktu kontrolnego SAC. Do białek tych należą m.in. BUB1 i MAD2L1. Hamują one aktywność kompleksu promującego anafazę do czasu uzyskania prawidłowego połączenia chromosomów z wrzecionem mitotycznym. Po 48 godz. hodowli zaobserwowano spadek ekspresji tych genów, co sugeruje, że nastąpił poślizg mitotyczny. Uważa się również, że jest on wynikiem zmienionej transkrypcyjnej regulacji genów, kodujących białka punktu kontrolnego G2/M oraz SAC [11].
Stwierdzono, że komórki pozbawione białek p53, p21 albo pRb i przejściowo zatrzymane w fazie M cyklu komórkowego na skutek uszkodzeń wrzeciona kariokinetycznego, po poślizgu mitotycznym wkraczają w fazę S, mimo że punkt kontrolny SAC działa poprawnie. Prowadzi to do poliploidyzacji materiału genetycznego. Zależny od białek p53 i pRb punkt kontrolny G1 jest zatem niezbędny w ochronie komórek przed poliploidalnością [85].
Inne mechanizmy działania docetakselu
Docetaksel oddziałuje na wiele struktur komórkowych i aktywuje szlaki przekaźnictwa sygnałów prowadzących do apoptozy. Działanie docetakselu na komórki wywołuje ich śmierć głównie poprzez szlak apoptotyczny, który może być niezależny od zatrzymania komórki w punktach kontrolnych cyklu, a związany z rozregulowaniem funkcji białek związanych z cyklem komórkowym, w tym szczególnie białka p34cdc-2, aktywacją szlaków kinazy fosforylującej N-terminalną część białka Jun-JNK (jun N-terminal kinase) i kinazy Raf-1 oraz indukcją białek rodziny Bcl-2 [46]. W wyniku aktywacji tego szlaku następuje proces fosforylacji białka Bcl-2, którego funkcją jest hamowanie apoptozy. Po przyłączeniu reszty fosforanowej białko ulega inaktywacji i komórka zostaje skierowana na szlak apoptozy. Stwierdzono, że docetaksel indukuje fosforylację białka Bcl-2 w większym stopniu niż paklitaksel, stąd obserwowane jest nasilenie procesu apoptozy w komórkach traktowanych docetakselem [27].
Oprócz działania cytotoksycznego taksany wykazują właściwości antyangiogenne [81]. Neowaskularyzacja tkanki nowotworowej, czyli powstawanie nowych naczyń włosowatych na bazie istniejących, jest niezbędnym etapem rozwoju nowotworu. Badania in vitro i in vivo z zastosowaniem docetakselu wykazały, że lek ten może blokować angiogenezę i tym samym prowadzić do zahamowania wzrostu guza oraz tworzenia przerzutów [32].
Podsumowanie
Istniejące obecnie schematy chemioterapii z zastosowaniem taksanów charakteryzują się wysokim ryzykiem wystąpienia działań niepożądanych. Ponadto pojawiająca się oporność komórek nowotworowych na stosowane leki powoduje, że chemioterapia jest często nieskuteczna [28,68]. Jednym z czynników powodujących oporność na taksany jest nadekspresja białek z rodziny ABC, związanych z ATP-zależną pompą usuwającą leki poza komórkę. Do białek tych zalicza się produkt genu MDR1 – glikoproteinę P (P-gp), białko MRP1 (multidrug resistance-associated protein 1) oraz białko BCRP (breast cancer resistance protein) znane również jako ABCG2 [9]. Oporność komórki na taksany może także wynikać ze zmian w strukturze tubuliny i mikrotubul, spowodowanych przez mutacje [88], zmienionej dynamiki mikrotubul [23] i różnic w ekspresji izotypów tubuliny [76]. U ludzi istnieje 7 izotypów β-tubuliny: I, II, III, IVA, IVB, V oraz VI, wykazujące różną ekspresję w zależności od tkanki. Stwierdzono, że izotypy I i IVB wykazują ekspresję konstytutywną, podczas gdy pozostałe izotypy są swoiste tkankowo [44]. Badania podstawowe i kliniczne sugerują, że ekspresja określonych izotypów tubulin jest związana z opornością komórek rakowych na taksany, a ekspresja izotypu III β-tubuliny może być prognostycznym czynnikiem w chemoterapii niektórych nowotworów np. niedrobnokomórkowego raka płuc (NSCLC – non-small cell lung carcinoma). Dlatego też stosowanie taksanów u pacjentów chorych na NSCLC z ekspresją III β-tubuliny jest nieskuteczne [75]. Wykazano ponadto, że izotypy I oraz IVA mogą się także przyczyniać do występowania oporności na taksany [58].
Proces nabywania przez komórki nowotworowe oporności na taksany I generacji spowodował rozwój badań związanych z nowymi pochodnymi. W zaawansowanej fazie badań klinicznych znajdują się dwie pochodne taksanów: larotaksel (XRP9881, RPR109881) i ortataksel (BAY59-8862, IDN5109, SB-T-101131), charakteryzujące się dużą cytotoksycznością wobec komórek opornych na działanie taksanów I generacji [73,95]. Prowadzone są również badania nad innymi taksanami II i III generacji, które charakteryzują się zwiększoną cytotoksycznością in vitro, wynikającą ze zmian w budowie cząsteczki [57]. Jedną z istniejących modyfikacji cząsteczki paklitakselu jest przyłączenie w pozycji 10 różnych grup funkcyjnych. Wśród wielu pochodnych, te z grupą benzoilową lub 4-metoksybenzoilową w pozycji 10 charakteryzują się najwyższą cytotoksycznością w linii komórkowej ludzkiego nowotworu piersi LCC6-MDR, wykazującej zwiększoną ekspresję białka P-gp. Pochodna z grupą 4-metoksyfenyloacetoksylową cechuje się natomiast największą spośród badanych związków cytotoksycznością w linii LCC6-WT (Pgp -) [55,56].
Inna modyfikacja polega na zmianie podstawników w pozycjach C-3′ i N-3′. Większość uzyskanych w ten sposób związków wykazuje wzmożoną cytotoksyczność zarówno w linii P-gp-, jak i w linii opornej o zwiększonej ekspresji P-gp. Na przykład najmniejszą wartość IC50 w linii LCC6-WT uzyskano dla pochodnej mającej podstawnik 2 metylopropenylowy 1 w pozycji C-3′ i resztę cyklopentyloksylową w pozycji N-3′. Dla linii LCC6-MDR był to związek z taką samą grupą funkcyjną w pozycji C-3′, z tym że w miejscu N-3′ podstawiony był cykloheksenyl 1. Do taksanów II generacji należą również fluorotaksoidy: C-3′-CF2 oraz C-3′ CF3. Cząsteczki te w większości charakteryzują się wzmożoną, w porównaniu z paklitakselem i docetakselem, cytotoksycznością zarówno wobec wrażliwych, jak i opornych nowotworowych linii komórkowych [55,56].
Zmienione właściwości taksanów nowej generacji wynikać mogą również z wprowadzenia grup funkcyjnych w pozycji meta podstawnika benzoilowego, znajdującego się w pozycji 2 pierścienia taksanowego. Stwierdzono m.in., że cząsteczka z podstawnikiem m-metoksylowym w tym miejscu oraz mająca grupę 4-metoksyfenyloacetoksylową przy węglu 10, ugrupowanie 1,1 dimetyloetenowe przy węglu 3′ oraz ugrupowanie t-butoksylowe w pozycji N-3′ wykazuje dużą cytotoksyczność zarówno wobec wrażliwych, jak i opornych linii komórkowych LCC6. Dodatkowo w obrębie tej grupy cząsteczek wyróżniono takie, które mogły pokonać barierę pompy związanej z białkiem Pgp. Związki te zaliczono do taksanów III generacji [56].
Inna grupa taksanów nowej generacji to C-sekotaksoidy, do których zaliczamy IDN 5390. Stwierdzono, że związek ten ze względu na swoją budowę charakteryzuje się 8-krotnie większą cytotoksycznością niż paklitaksel wobec nowotworowych linii komórkowych. Zwiększona cytotoksyczność wynika z odmiennego sposobu oddziaływania z izotypem III β-tubuliny [19].
Synteza wielu pochodnych taksoidów II i III generacji, aktywnych w komórkach rakowych, które wykazują oporność wielolekową (MDR) (w tym także i na taksany I generacji) budzi nadzieję na bardziej efektywne zastosowanie nowych inhibitorów mitozy w onkologii klinicznej.
Podziękowania
Autorzy składają serdeczne podziękowania Panu Profesorowi Jamesowi P. Snyderowi za dostarczenie ryciny przedstawiającej miejsce łączenia się taksanu z podjednostką b-tubuliny oraz Panu Profesorowi Kennethowi H. Downingowi za wykonanie i dostarczenie ryciny pokazującej strukturę tubuliny
Acknowledgements
The authors would like to thank professor James P. Snyder for providing the figure, which depicts connection of taxanes with b-tubulin and professor Kenneth H. Downing for making and providing the figure of new, refined tubulin structure.
PIŚMIENNICTWO
[1] Abbrederis K., Lorenzen S., von Weikersthal L.F., Vehling-Kaiser U., Schuster T., Rothling N., Peschel C., Lordick F.: Weekly docetaxel monotherapy for advanced gastric or esophagogastric junction cancer. Results of a phase II study in elderly patients or patients with impaired performance status. Crit. Rev. Oncol. Hematol., 2008; 66: 84-90
[PubMed]
[2] Aihara T., Kim Y., Takatsuka Y.: Phase II study of weekly docetaxel in patients with metastatic breast cancer. Ann. Oncol., 2002; 13: 286-292
[PubMed] [Full Text HTML] [Full Text PDF]
[3] Alcaraz A.A., Mehta A.K., Johnson S.A., Snyder J.P.: The T-taxol conformation. J. Med. Chem., 2006; 49: 2478-2488
[PubMed]
[4] Amos L.A.: Focusing-in on microtubules. Curr. Opin. Struct. Biol., 2000; 10: 236-241
[PubMed]
[5] Andreu J.M., Bordas J., Diaz J.F., Garcia de Ancos J., Gil R., Medrano F.J., Nogales E., Pantos E., Towns-Andrews E.: Low resolution structure of microtubules in solution. Synchrotron X-ray scattering and electron microscopy of taxol-induced microtubules assembled from purified tubulin in comparison with glycerol and MAP-induced microtubules. J. Mol. Biol., 1992; 226: 169-184
[PubMed]
[6] Baker S.D., Zhao M., Lee C.K., Verweij J., Zabelina Y., Brahmer J.R., Wolff A.C., Sparreboom A., Carducci M.A.: Comparative pharmacokinetics of weekly and every-three-weeks docetaxel. Clin. Cancer Res., 2004; 10: 1976-1983
[PubMed] [Full Text HTML] [Full Text PDF]
[7] Biffi R., Fazio N., Luca F., Chiappa A., Andreoni B., Zampino M.G., Roth A., Schuller J.C., Fiori G., Orsi F., Bonomo G., Crosta C., Huber O.: Surgical outcome after docetaxel-based neoadjuvant chemotherapy in locally-advanced gastric cancer. World J. Gastroenterol., 2010; 16: 868-874
[PubMed] [Full Text HTML] [Full Text PDF]
[8] Bodnar L., Wcisło G., Miedzińska-Maciejewska M., Szczylik C.: Docetaksel i paklitaksel: porównanie ich budowy, farmakologii oraz mechanizmów oporności. Współczesna Onkologia, 2004; 8: 435-446
[9] Brooks T.A., Minderman H., O’Loughlin K.L., Pera P., Ojima I., Baer M.R., Bernacki R.J.: Taxane-based reversal agents modulate drug resistance mediated by P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Mol. Cancer Ther., 2003; 2: 1195-1205
[PubMed] [Full Text HTML] [Full Text PDF]
[10] Chau M., Jennewein S., Walker K., Croteau R.: Taxol biosynthesis: molecular cloning and characterization of a cytochrome P450 taxoid 7β-hydroxylase. Chem. Biol., 2004; 11: 663-672
[PubMed]
[11] Chen J.G., Yang C.P., Cammer M., Horwitz S.B.: Gene expression and mitotic exit induced by microtubule-stabilizing drugs. Cancer Res., 2003; 63: 7891-7899
[PubMed] [Full Text HTML] [Full Text PDF]
[12] Chrétien D., Wade R.H.: New data on the microtubule surface lattice. Biol. Cell, 1991; 71: 161-74
[PubMed]
[13] Cortes J.E., Pazdur R.: Docetaxel. J. Clin. Oncol., 1995; 13: 2643-2655
[PubMed]
[14] Cresteil T., Monsarrat B., Dubois J., Sonnier M., Alvinerie P., Gueritte F.: Regioselective metabolism of taxoids by human CYP3A4 and 2C8: structure-activity relationship. Drug Metab. Dispos., 2002; 30: 438-445
[PubMed] [Full Text HTML] [Full Text PDF]
[15] Downing K.H., Nogales E.: Crystallographic structure of tubulin: implications for dynamics and drug binding. Cell Struct. Funct., 1999; 24: 269-275
[PubMed] [Full Text PDF]
[16] Dutcher S.K.: The tubulin fraternity: α to ε. Curr. Opin. Cell Biol., 2001; 13: 49-54
[PubMed]
[17] Dye R.B., Flicker P.F., Lien D.Y., Williams R.C. Jr.: End-stabilized microtubules observed in vitro: stability, subunit, interchange, and breakage. Cell Motil. Cytoskeleton, 1992; 21: 171-186
[PubMed]
[18] Estévez L.G., Cuevas J.M., Antón A., Florián J., López-Vega J.M., Velasco A., Lobo F., Herrero A., Fortes J.: Weekly docetaxel as neoadjuvant chemotherapy for stage II and III breast cancer: efficacy and correlation with biological markers in a phase II, multicenter study. Clin. Cancer Res., 2003; 9: 686-692
[PubMed] [Full Text HTML] [Full Text PDF]
[19] Ferlini C., Raspaglio G., Mozzetti S., Cicchillitti L., Filippetti F., Gallo D., Fattorusso C., Campiani G., Scambia G.: The seco-taxane IDN5390 is able to target class III β-tubulin and to overcome paclitaxel resistance. Cancer Res., 2005; 65: 2397-2405
[PubMed] [Full Text HTML] [Full Text PDF]
[20] Fuller S.D., Gowen B.E., Reinsch S., Sawyer A., Buendia B., Wepf R., Karsenti E.: The core of the mammalian centriole contains γ-tubulin. Curr. Biol., 1995; 5: 1384-1393
[PubMed]
[21] Giannakakou P., Sackett D.L., Kang Y.K., Zhan Z., Buters J.T., Fojo T., Poruchynsky M.S.: Paclitaxel-resistant human ovarian cancer cells have mutant β-tubulins that exhibit impaired paclitaxel-driven polymerization. J. Biol. Chem., 1997; 272: 17118-17125
[PubMed] [Full Text HTML] [Full Text PDF]
[22] Goble S., Bear H.D.: Emerging role of taxanes in adjuvant and neoadjuvant therapy for breast cancer: the potential and the questions. Surg. Clin. North Am., 2003; 83: 943-971
[PubMed]
[23] Gonçalves A., Braguer D., Kamath K., Martello L., Briand C., Horwitz S., Wilson L., Jordan M.A.: Resistance to taxol in lung cancer cells associated with increased microtubule dynamics. Proc. Natl. Acad. Sci. USA, 2001; 98: 11737-11742
[PubMed] [Full Text HTML] [Full Text PDF]
[24] Goodman B., Zheng Y.: Mitotic spindle morphogenesis: Ran on the microtubule cytoskeleton and beyond. Biochem. Soc. Trans., 2006; 34: 716-721
[PubMed] [Full Text HTML] [Full Text PDF]
[25] Gunawardane R.N., Lizarraga S.B., Wiese C., Wilde A., Zheng Y.: γ-Tubulin complexes and their role in microtubule nucleation. Curr. Top. Dev. Biol., 2000; 49: 55-73
[PubMed]
[26] Hainsworth J.D., Burris H.A.3rd, Yardley D.A., Bradof J.E., Grimaldi M., Kalman L.A., Sullivan T., Baker M., Erland J.B., Greco F.A.: Weekly docetaxel in the treatment of elderly patients with advanced breast cancer: a Minnie Pearl Cancer Research Network phase II trial. J. Clin. Oncol., 2001; 19: 3500-3505
[PubMed] [Full Text HTML] [Full Text PDF]
[27] Haldar S., Basu A., Croce C.M.: Bc12 is the guardian of microtubule integrity. Cancer Res., 1997; 57: 229-233
[PubMed] [Full Text PDF]
[28] Hayashi Y., Kuriyama H., Umezu H., Tanaka J., Yoshimasu T., Furukawa T., Tanaka H., Kagamu H., Gejyo F., Yoshizawa H.: Class III β-tubulin expression in tumor cells is correlated with resistance to docetaxel in patients with completely resected non-small-cell lung cancer. Intern. Med., 2009; 48: 203-208
[PubMed] [Full Text PDF]
[29] Hernández-Vargas H., Palacios J., Moreno-Bueno G.: Telling cells how to die: docetaxel therapy in cancer cell lines. Cell Cycle, 2007; 6: 780-783
[PubMed] [Full Text PDF]
[30] Hollenbeck P.: Cytoskeleton: microtubules get the signal. Curr. Biol., 2001; 11: R820-R823
[PubMed]
[31] Honore S., Pasquier E., Braguer D.: Understanding microtubule dynamics for improved cancer therapy. Cell. Mol. Life Sci., 2005; 62: 3039-3056
[PubMed] [Full Text PDF]
[32] Hotchkiss K.A., Ashton A.W., Mahmood R., Russell R.G., Sparano J.A., Schwartz E.L.: Inhibition of endothelial cell function in vitro and angiogenesis in vivo by docetaxel (Taxotere): association with impaired repositioning of the microtubule organizing center. Mol. Cancer Ther., 2002; 1: 1191-1200
[PubMed] [Full Text HTML] [Full Text PDF]
[33] Hudis C., McArthur H., Dang C.: Current status of the taxanes as adjuvant therapy for breast cancer. Breast, 2007; 16 (suppl. 2): S132-S135
[PubMed]
[34] Jánosi I.M., Chrétien D., Flyvbjerg H.: Structural microtubule cap: stability, catastrophe, rescue, and third state. Biophys. J., 2002; 83: 1317-1330
[PubMed] [Full Text PDF]
[35] Jones S.E., Erban J., Overmoyer B., Budd G.T., Hutchins L., Lower E., Laufman L., Sundaram S., Urba W.J., Pritchard K.I., Mennel R., Richards D., Olsen S., Meyers M.L., Ravdin P.M.: Randomized phase III study of docetaxel compared with paclitaxel in metastatic breast cancer. J. Clin. Oncol., 2005; 23: 5542-5551
[PubMed] [Full Text HTML] [Full Text PDF]
[36] Jordan M.A., Wilson L.: Microtubules as a target for anticancer drugs. Nat. Rev. Cancer, 2004; 4: 253-265
[PubMed]
[37] Keskin O., Durell S.R., Bahar I., Jernigan R.L., Covell D.G.: Relating molecular flexibility to function: a case study of tubulin. Biophys. J., 2002; 83: 663-680
[PubMed] [Full Text PDF]
[38] King K.M., Lupichuk S., Baig L., Webster M., Basi S., Whyte D., Rix S.: Optimal use of taxanes in metastatic breast cancer. Curr. Oncol., 2009; 16: 8-20
[PubMed] [Full Text PDF]
[39] Li H., DeRosier D.J., Nicholson W.V., Nogales E., Downing K.H.: Microtubule structure at 8A resolution. Structure, 2002; 10: 1317-1328
[PubMed]
[40] Liu S.M., Magnusson K.E., Sundqvist T.: Microtubules are involved in transport of macromolecules by vesicles in cultured bovine aortic endothelial cells. J. Cell. Physiol., 1993; 156: 311-316
[PubMed]
[41] Lo S.S, Khorana A.A., Javle M., Simon S., Kiefer G., Rajasenan K., Wang H., Hantel A., Shayne M., Hwang J., Schmotzer A., Ramanathan R.K.: A phase II study of weekly docetaxel in combination with capecitabine in advanced gastric and gastroesophageal adenocarcinomas. Oncology, 2010; 78: 125-129
[PubMed]
[42] Löwe J., Amos L.A.: Crystal structure of the bacterial cell-division protein FtsZ. Nature, 1998; 391: 203-206
[PubMed]
[43] Löwe J., Li H., Downing K.H., Nogales E.: Refined structure of α β-tubulin at 3.5 A resolution. J. Mol. Biol., 2001; 313: 1045-1057
[PubMed]
[44] McGrogan B.T., Gilmartin B., Carney D.N., McCann A.: Taxanes, microtubules and chemoresistant breast cancer. Biochim. Biophys. Acta, 2008; 1785: 96-132
[PubMed]
[45] Mey U., Gorschlüter M., Ziske C., Kleinschmidt R., Glasmacher A., Schmidt-Wolf I.G.: Weekly docetaxel in patients with pretreated metastatic breast cancer: a phase II trial. Anticancer Drugs, 2003; 14: 233-238
[PubMed]
[46] Mhaidat N.M., Zhang X.D., Jiang C.C., Hersey P.: Docetaxel-induced apoptosis of human melanoma is mediated by activation of c-Jun NH2-terminal kinase and inhibited by the mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway. Clin. Cancer Res., 2007; 13: 1308-1314
[PubMed] [Full Text HTML] [Full Text PDF]
[47] Mitchison T.J.: Localization of an exchangeable GTP binding site at the plus end of microtubules. Science, 1993; 261: 1044-1047
[PubMed]
[48] Miyamoto D.T., Perlman Z.E., Mitchison T.J., Shirasu-Hiza M.: Dynamics of the mitotic spindle – potential therapeutic targets. Prog. Cell Cycle Res., 2003; 5: 349-360
[PubMed]
[49] Mollinedo F., Gajate C.: Microtubules, microtubule-interfering agents and apoptosis. Apoptosis, 2003; 8: 413-450
[PubMed]
[50] Monsarrat B., Thoison O., Dubois J., Cresteil T.: Molecular determinant of regioselective hydroxylation of docetaxel by CYP3A4. Mol. Cell. Pharmacol., 2009; 1: 76-84
[Abstract] [Full Text PDF]
[51] Moritz M., Braunfeld M.B., Guénebaut V., Heuser J., Agard D.A.: Structure of the γ-tubulin ring complex: a template for microtubule nucleation. Nat. Cell Biol., 2000; 2: 365-370
[PubMed]
[52] Morse D.L., Gray H., Payne C.M., Gillies R.J.: Docetaxel induces cell death through mitotic catastrophe in human breast cancer cells. Mol. Cancer Ther., 2005; 4: 1495-1504
[PubMed] [Full Text HTML] [Full Text PDF]
[53] Nogales E., Whittaker M., Milligan R.A., Downing K.H.: High-resolution model of the microtubule. Cell, 1999; 96: 79-88
[PubMed]
[54] Ohi R., Gould K.L.: Regulating the onset of mitosis. Curr. Opin. Cell Biol., 1999; 11: 267-273
[PubMed]
[55] Ojima I., Chen J., Sun L., Borella C.P., Wang T., Miller M.L., Lin S., Geng X., Kuznetsova L., Qu C., Gallager D., Zhao X., Zanardi I., Xia S., Horwitz S.B., Mallen-St. Clair J., Guerriero J.L., Bar-Sagi D., Veith J.M., Pera P., Bernacki R.J.: Design, synthesis, and biological evaluation of new-generation taxoids. J. Med. Chem., 2008; 51: 3203-3221
[PubMed]
[56] Ojima I., Das M.: Recent advances in the chemistry and biology of new generation taxoids. J. Nat. Prod., 2009; 72: 554-565
[PubMed] [Full Text PDF]
[57] Ojima I., Slater J.C., Michaud E., Kuduk S.D., Bounaud P.Y., Vrignaud P., Bissery M.C., Veith J.M., Pera P., Bernacki R.J.: Syntheses and structure-activity relationships of the second-generation antitumor taxoids: exceptional activity against drug-resistant cancer cells. J. Med. Chem., 1996; 39: 3889-3896
[PubMed]
[58] Orr G.A., Verdier-Pinard P., McDaid H., Horwitz S.B.: Mechanisms of taxol resistance related to microtubules. Oncogene, 2003; 22: 7280-7295
[PubMed] [Full Text HTML] [Full Text PDF]
[59] Otová B., Václaviková R., Danielová V., Holubová J., Ehrlichová M., Horský S., Soucek P., Simek P., Gut I.: Effects of paclitaxel, docetaxel and their combinations on subcutaneous lymphomas in inbred Sprague-Dawley/Cub rats. Eur. J. Pharm. Sci., 2006; 29: 442-450
[PubMed]
[60] Paoletti A., Giocanti N., Favaudon V., Bornens M.: Pulse treatment of interphasic HeLa cells with nanomolar doses of docetaxel affects centrosome organization and leads to catastrophic exit of mitosis. J. Cell Sci., 1997; 110: 2403-2415
[PubMed] [Full Text PDF]
[61] Pasquier E., Kavallaris M.: Microtubules: a dynamic target in cancer therapy. IUBMB Life, 2008; 60: 165-170
[PubMed] [Full Text HTML] [Full Text PDF]
[62] Radulescu A.E., Cleveland D.W.: NuMA after 30 years: the matrix revisited. Trends Cell Biol., 2010; 20: 214-222
[PubMed]
[63] Rao S., He L., Chakravarty S., Ojima I., Orr G.A., Horwitz S.B.: Characterization of the Taxol binding site on the microtubule. Identification of Arg282 in β-tubulin as the site of photoincorporation of a 7-benzophenone analogue of Taxol. J. Biol. Chem., 1999; 274: 37990-37994
[PubMed] [Full Text HTML] [Full Text PDF]
[64] Rieder C.L., Alexander S.P.: Kinetochores are transported poleward along a single astral microtubule during chromosome attachment to the spindle in newt lung cells. J. Cell Biol., 1990; 110: 81-95
[PubMed] [Full Text PDF]
[65] Rieder C.L., Maiato H.: Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell, 2004; 7: 637-651
[PubMed]
[66] Riou J.F., Naudin A., Lavelle F.: Effects of Taxotere on murine and human tumor cell lines. Biochem. Biophys. Res. Commun., 1992; 187: 164-170
[PubMed]
[67] Riou J.F., Petitgenet O., Combeau C., Lavelle F.: Cellular uptake and efflux of docetaxel (Taxotere) and paclitaxel (Taxol) in P388 cell line. Proc. Am. Assoc. Cancer Res., 1994; 35: 385, Abstr. 2292
[68] Roglio I., Bianchi R., Camozzi F., Carozzi V., Cervellini I., Crippa D., Lauria G., Cavaletti G., Melcangi R.C.: Docetaxel-induced peripheral neuropathy: protective effects of dihydroprogesterone and progesterone in an experimental model. J. Peripher. Nerv. Syst., 2009; 14: 36-44
[PubMed]
[69] Rowinsky E.K.: The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu. Rev. Med., 1997; 48: 353-374
[PubMed]
[70] Royer I., Monsarrat B., Sonnier M., Wright M., Cresteil T.: Metabolism of docetaxel by human cytochromes P450: interactions with paclitaxel and other antineoplastic drugs. Cancer Res., 1996; 56: 58-65
[PubMed] [Full Text PDF]
[71] Sakaushi S., Nishida K., Minamikawa H., Fukada T., Oka S., Sugimoto K.: Live imaging of spindle pole disorganization in docetaxel-treated multicolor cells. Biochem. Biophys. Res. Commun., 2007; 357: 655-660
[PubMed]
[72] Salminen E., Bergman M., Huhtala S., Ekholm E.: Docetaxel: standard recommended dose of 100 mg/m2 is effective but not feasible for some metastatic breast cancer patients heavily pretreated with chemotherapy – A phase II single-center study. J. Clin. Oncol., 1999; 17: 1127
[PubMed] [Full Text HTML] [Full Text PDF]
[73] Sano D., Matsuda H., Ishiguro Y., Nishimura G., Kawakami M., Tsukuda M.: Antitumor effects of IDN5109 on head and neck squamous cell carcinoma. Oncol. Rep., 2006; 15: 329-334
[PubMed] [Full Text PDF]
[74] Schmidt M., Bastians H.: Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist. Updat., 2007; 10: 162-181
[PubMed]
[75] Seve P., Mackey J., Isaac S., Trédan O., Souquet P.J., Pérol M., Lai R., Voloch A., Dumontet C.: Class III β-tubulin expression in tumor cells predicts response and outcome in patients with non-small cell lung cancer receiving paclitaxel. Mol. Cancer Ther., 2005; 4: 2001-2007
[PubMed] [Full Text HTML] [Full Text PDF]
[76] Shalli K., Brown I., Heys S.D., Schofield A.C.: Alterations of β-tubulin isotypes in breast cancer cells resistant to docetaxel. FASEB J., 2005; 19: 1299-1301
[PubMed] [Full Text PDF]
[77] Snyder J.P., Nettles J.H., Cornett B., Downing K.H., Nogales E.: The binding conformation of Taxol in β-tubulin: a model based on electron crystallographic density. Proc. Natl. Acad. Sci. USA, 2001; 98: 5312-5316
[PubMed] [Full Text HTML] [Full Text PDF]
[78] Vaclavikova R., Soucek P., Svobodova L., Anzenbacher P., Simek P., Guengerich F.P., Gut I.: Different in vitro metabolism of paclitaxel and docetaxel in humans, rats, pigs, and minipigs. Drug Metab. Dispos., 2004; 32: 666-674
[PubMed] [Full Text HTML] [Full Text PDF]
[79] Tao W.: The mitotic checkpoint in cancer therapy. Cell Cycle, 2005; 4: 1495-1499
[PubMed] [Full Text PDF]
[80] Ueno H., Yasunaga T., Shingyoji C., Hirose K.: Dynein pulls microtubules without rotating its stalk. Proc. Natl. Acad. Sci. USA, 2008; 105: 19702-19707
[PubMed] [Full Text HTML] [Full Text PDF]
[81] Vacca A., Ribatti D., Iurlaro M., Merchionne F., Nico B., Ria R., Dammacco F.: Docetaxel versus paclitaxel for antiangiogenesis. J. Hematother. Stem Cell Res., 2002; 11: 103-118
[PubMed]
[82] Valerio M.R., Russo A., Latteri M.A., Modica G., Gulotta G., Armata M.G., Bajardi E., Cicero G., Pantuso G., Grassi N., Agosta G., Gebbia N.: Weekly docetaxel as II line therapy in non-small cell lung cancer: an interim analysis of a phase II study. Lung Cancer, 2001; 34 (Suppl. 4): S31-S35
[PubMed]
[83] van Herwaarden A.E., van Waterschoot R.A., Schinkel A.H.: How important is intestinal cytochrome P-450 3A metabolism? Trends Pharmacol. Sci., 2009; 30: 223-227
[PubMed]
[84] Vobořilová J., Němcová-Fürstová V., Neubauerová J., Ojima I., Zanardi I., Gut I., Kovář J.: Cell death induced by novel fluorinated taxanes in drug-sensitive and drug-resistant cancer cells. Invest. New Drugs, 2011 (w druku)
[PubMed]
[85] Vogel C., Kienitz A., Hofmann I., Müller R., Bastians H.: Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene, 2004; 23: 6845-6853
[PubMed] [Full Text HTML] [Full Text PDF]
[86] Wagner P., Wang B., Clark E., Lee H., Rouzier R., Pusztai L.: Microtubule associated protein (MAP)-τ: a novel mediator of paclitaxel sensitivity in vitro and in vivo. Cell Cycle, 2005; 4: 1149-1152
[PubMed] [Full Text PDF]
[87] Wang H.W., Nogales E.: Nucleotide-dependent bending flexibility of tubulin regulates microtubule assembly. Nature, 2005; 435: 911-915
[PubMed] [Full Text PDF]
[88] Wang Y., O’Brate A., Zhou W., Giannakakou P.: Resistance to microtubule-stabilizing drugs involves two events: β-tubulin mutation in one allele followed by loss of the second allele. Cell Cycle, 2005; 4: 1847-1853
[PubMed] [Full Text PDF]
[89] Warner S.L, Gray P.J., Von Hoff D.D.: Tubulin-associated drug targets: Aurora kinases, Polo-like kinases, and others. Semin. Oncol., 2006; 33: 436-448
[PubMed]
[90] Weaver B.A., Cleveland D.W.: Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell, 2005; 8: 7-12
[PubMed]
[91] Welte M.A.: Bidirectional transport along microtubules. Curr. Biol., 2004; 14; R525-R537
[PubMed]
[92] Xiao H., Verdier-Pinard P., Fernandez-Fuentes N., Burd B., Angeletti R., Fiser A., Horwitz S.B., Orr G.A.: Insights into the mechanism of microtubule stabilization by Taxol. Proc. Natl. Acad. Sci. USA, 2006; 103: 10166-10173
[PubMed] [Full Text HTML] [Full Text PDF]
[93] Yang Y., Alcaraz A.A., Snyder J.P.: The tubulin-bound conformation of paclitaxel: T-Taxol vs „PTX-NY”. J. Nat. Prod., 2009; 72: 422-429
[PubMed]
[94] Yvon A.M., Wadsworth P.: Non-centrosomal microtubule formation and measurement of minus end microtubule dynamics in A498 cells. J. Cell Sci., 1997; 110: 2391-2401
[PubMed] [Full Text PDF]
[95] Zatloukal P., Gervais R., Vansteenkiste J., Bosquee L., Sessa C., Brain E., Dansin E., Urban T., Dohollou N., Besenval M., Quoix E.: Randomized multicenter phase II study of larotaxel (XRP9881) in combination with cisplatin or gemcitabine as first-line chemotherapy in nonirradiable stage IIIB or stage IV non-small cell lung cancer. J. Thorac. Oncol., 2008; 3: 894-901
[PubMed]
[96] Zhou J., Giannakakou P.: Targeting microtubules for cancer chemotherapy. Curr. Med. Chem. Anticancer Agents, 2005; 5: 65-71
[PubMed]
Autorzy deklarują brak potencjalnych konfliktów interesów.