
Molekularne podstawy dziedzicznej hemochromatozy
Tomasz Romanowski 1 , Katarzyna Sikorska , Krzysztof Piotr BielawskiStreszczenie
Dziedziczna hemochromatoza (HH) jest genetyczną chorobą metaboliczną. Charakteryzuje się nadmierną absorpcją żelaza z pokarmu i progresywną akumulacją tego metalu w komórkach wielu narządów. Na homeostazę żelaza w organizmie człowieka składa się wiele skomplikowanych procesów, z których część nie została jeszcze poznana. Badania genetyczne pacjentów dotkniętych HH przyczyniły się w ciągu ostatnich lat do odkrycia wielu nowych białek i mechanizmów wpływających na wchłanianie, transport, magazynowanie i wydalanie żelaza. Dzięki nim zauważono również jak bardzo złożone jest to schorzenie i jak mutacje mogą modyfikować jego obraz kliniczny. W pracy przedstawiono aktualną wiedzę na temat mechanizmów biorących udział w metabolizmie żelaza, klasyfikację i typy dziedzicznej hemochromatozy oraz mutacje wywołujące tę chorobę.
Słowa kluczowe:hemochromatoza • żelazo • HFE • HAMP • HJV • TFR2 • SLC40A1
Summary
Hereditary hemochromatosis (HH) is a genetic metabolic disease characterized by increased intestinal iron absorption and progressive iron loading in the cells of various organs. Human body iron homeostasis involves a number of complicated processes, some of which are not identified yet. Genetic analysis of patients affected by HH recently led to the discovery of many novel proteins and mechanisms that can influence the uptake, transport, storage, and excretion of iron. It also showed that hemochromatosis is a very complex disease and that the type of mutation can influence its clinical manifestation. This review presents the current knowledge about the mechanisms of iron metabolism and describes the types of hereditary hemochromatosis and the mutations which induce the disease.
Key words:hemochromatosis • iron • HFE • HAMP • HJV • TFR2 • SLC40A1
Wykaz skrótów:b
2M – b
2-mikroglobulina; Dcytb – cytochrom b dwunastnicy (duodenal cytochrome b); DMT1 – transporter metali dwuwartościowych 1 (divalent metal transporter 1); IRE – element wrażliwy na żelazo (iron responsive element); HAMP – gen kodujący hepcydynę; HH – dziedziczna hemochromatoza (hereditary hemochromatosis); HJV – gen kodujący hemojuwelinę; JH – młodzieńcza hemochromatoza (juvenile hemochromatosis); LEAP-1 – wątrobowy peptyd antybakteryjny 1 (liver expressed antimicrobial peptide-1); MHC – główny układ zgodności tkankowej (major histocompatibility complex); ROS – reaktywne formy tlenu (reactive oxygene species); SLC40A1 – gen kodujący ferroportynę; Tf – transferyna (transferin); Tf-Fe – kompleks transferyny i żelaza; TfR – receptor transferyny; TFR2 – gen kodujący receptor transferyny 2.
Dziedziczna (wrodzona) hemochromatoza (hereditary hemochromatosis – HH) jest jedną z częstszych chorób genetycznych wśród przedstawicieli rasy białej. Schorzenie to dotyka 0,25–0,5% mieszkańców Europy Północnej [5]. Jest to choroba metaboliczna, w której dochodzi do nadmiernego wchłaniania żelaza z pokarmu i odkładania się tego pierwiastka w miąższowych komórkach wątroby, serca, trzustki i gruczołów dokrewnych. Przełom w badaniach nad HH nastąpił w 1996 r., kiedy zidentyfikowano gen hemochromatozy (HFE) [31]. Odtąd zaczęto wykrywać także inne geny, których produkty odgrywają zasadniczą rolę w regulacji wchłaniania, transporcie i wewnątrzustrojowej dystrybucji żelaza. Mutacje tych genów sprzyjają wystąpieniu istotnych zaburzeń metabolizmu żelaza, co prowadzi do stopniowego rozwoju choroby – hemochromatozy.
ŻELAZO W ORGANIZMIE CZŁOWIEKA
Żelazo jest niezbędnym do życia składnikiem prawie wszystkich organizmów. Wiele podstawowych procesów metabolicznych, takich jak synteza DNA, RNA, transport tlenu, elektronów, przebiega w oparciu o reakcje enzymatyczne z udziałem oksydaz, katalaz, peroksydaz, cytochromów, reduktaz rybonukleotydowych, akonitaz, których niezbędnym kofaktorem są jony żelaza [11,92,110]. Regulując transkrypcję kilku genów (kinezy białkowej C-b
izozymu 5 kwaśnej fosfatazy, p21) jony żelaza wpływają na cykl komórkowy, różnicowanie, proliferację [11]. Ich obecność jest także niezbędna do tworzenia osłonki mielinowej oraz wypustek protoplazmatycznych neuronów [35].
Funkcja żelaza w przemianach metabolicznych wynika z jego własności chemicznych. Występuje ono w dwóch stanach utlenienia, w postaci jonów Fe+2 i Fe+3, dzięki czemu może być akceptorem i donorem elektronów. Można to przedstawić za pomocą reakcji Fentona [110]:

Udział żelaza w wyżej wymienionych reakcjach pozwala na regulację wielu przemian metabolicznych na poziomie komórkowym. Zarazem jednak nadmiar tego metalu sprzyja wytwarzaniu reaktywnych form tlenu (reactive oxygene species – ROS), które działają destruktywnie na komórki. Powstają wysoko toksyczne rodniki wodorotlenowe (OH–) i ponadtlenkowe (•O2), które mają zdolność łatwego reagowania z większością zawartych w komórkach cząstek [70]. Powodują one uszkodzenie DNA, upośledzają mechanizmy syntezy białek, lipidów, węglowodanów, indukują proteazy, wpływają na proliferację, w niektórych przypadkach powodują nawet śmierć komórek [40]. W odpowiedzi na te procesy w komórkach wykształciły się mechanizmy mające chronić przed destrukcją, dzięki którym jony żelaza są wiązane, transportowane i magazynowane w nietoksycznych, rozpuszczalnych postaciach.
Ilość żelaza w organizmie dorosłego człowieka to około 40 mg na kilogram masy ciała [3]. Jego znaczna część (60–70%) pozostaje związana w hemoglobinie krążących we krwi erytrocytów. Kolejne 10% jest obecne w postaci mioglobin, cytochromów i różnych enzymów. Pozostałe 20–30% żelaza jest gromadzone w postaci ferrytyny i hemosyderyny w hepatocytach i siateczkowo-śródbłonkowych makrofagach [22].
Transferyna stanowi najważniejszy nośnik żelaza, mimo iż pozostaje z nią związany tylko 1% (około 4 mg) całych zasobów wewnątrzustrojowych tego pierwiastka. W ciągu dnia transferyna transportuje około 25 mg żelaza, z czego 80% przenoszone jest do szpiku kostnego, gdzie w retikulocytach (komórkach prekursorowych erytrocytów) zachodzi synteza hemoglobiny [22]. Możemy w tym przypadku mówić o obiegu zamkniętym. Większość użytego w tym procesie żelaza pochodzi z rozkładu starych erytrocytów. Śródbłonkowe makrofagi trawią w śledzionie i wątrobie stare czerwone krwinki i uwalniają zawarte w hemoglobinie żelazo do krwiobiegu. Stąd transferyna przenosi je do komórek szpiku kostnego, gdzie za pomocą ferrochelatazy zostaje ono związane z protoporfiryną IX, tworząc cząsteczki hemu [14,69]. Hem łącząc się z podjednostkami białkowymi tworzy hemoglobinę, najważniejszy nośnik tlenu.
Jest kilka fizjologicznych mechanizmów wydalania żelaza z organizmu. Usuwane jest ono razem z żółcią, moczem oraz z łuszczącymi się komórkami skóry i jelit, co stanowi jedynie 1 mg na dzień u dorosłego człowieka [3]. Większe jego straty możliwe są u kobiet w wyniku comiesięcznej utraty żelaza w czasie menstruacji oraz w okresie ciąży, porodu i laktacji [113]. Utrzymanie stałego poziomu żelaza możliwe jest dzięki wchłanianiu tego pierwiastka z pokarmu. Jego absorpcja w organizmie człowieka wynosi 1–4 mg na dzień i odbywa się w jelicie cienkim.
METABOLIZM ŻELAZA
Kontrola ilości żelaza w ustroju odbywa się w dużo większym zakresie za pomocą monitorowania jego absorpcji niż zmiennej ekskrecji. W organizmie człowieka za uzupełnianie zawartości tego pierwiastka odpowiedzialne są enterocyty nabłonka dwunastnicy. Komórki te wykazują polaryzację. Ich warstwa szczytowa, zwrócona w stronę światła jelita, wyspecjalizowana jest w transporcie hemu i jonów żelaza do wnętrza komórek. Istnieją co najmniej trzy szlaki tego transportu [98]. Najbardziej znany jest związany z nośnikiem metali dwuwartościowych (divalent metal transporter – DMT1; nazwy zastępcze: Nramp2, DCT1). DMT1 jest protonowym symporterem, przenoszącym kationy żelaza oraz inne dwuwartościowe metale ze światła jelita do wnętrza enterocytów [39]. Niezbędna jest tutaj obecność białek pomocniczych, takich jak cytochrom b (duodenal cytochrome b – Dcytb), który redukuje zawarte w pokarmie żelazo [71]. Drugim bardzo znaczącym, choć dotąd słabo opisanym, mechanizmem wchłaniania tego pierwiastka jest absorpcja cząsteczek hemu. Znany jest także cykl mucyna–integryna–mobilferryna, który również pozwala na wnikanie jonów żelaza do komórek nabłonkowych jelita [22].
Przez podstawną część nabłonka odbywa się natomiast transport znajdującego się już w erytrocytach żelaza do naczyń krwionośnych. Uczestniczy w tym ferroportyna [1] i ułatwiająca przenikanie hefajstyna [105]. Ferroportyna, nazywana również bazolateralnym transporterem żelaza (Ireg1/MTP1), jest dużym białkiem transmembranowym. Poza dwunastnicą występuje ona w komórkach wątroby, śledziony, nerek i w mających cechy makrofagów, komórkach Kupffera [1]. W procesie usuwania żelaza z komórek współpracuje z nią hefajstyna, wykazująca dużą homologię z obecną w osoczu ceruloplazminą [114]. To ostatnie białko jest miedziową ferroksydazą, enzymem niezbędnym do wymiany zmagazynowanego żelaza między wątrobą, systemem śródbłonkowym i krwią. Przeanalizowanie struktury hefajstyny pozwala stwierdzić, że za aktywność tego białka odpowiadają także atomy miedzi. Miedź utlenia żelazo z Fe+2 do Fe+3, co jest warunkiem jego transferu z enterocytów do surowicy.
W płynach ustrojowych utlenione żelazo wychwycone zostaje przez transferynę (Tf). Białko to transportuje żelazo do większości komórek w organizmie. Transferyna syntetyzowana jest głównie w wątrobie [64] i występuje w trzech postaciach: niezwiązanej z żelazem (apotransferyna) i związanej z jednym lub dwoma atomami żelaza (transferyna odpowiednio monoferryczna lub dwuferryczna). Przewaga ilości jednej z tych postaci zależy od stężenia żelaza w surowicy [47]. Związane z transferyną żelazo (Tf-Fe) przechodzi pierwotnie przez system wrotny wątroby, która jest głównym rezerwuarem tego pierwiastka w ustroju. Stamtąd kierowane jest do pozostałych organów i tkanek.
Kompleks Tf-Fe pobierany jest przez komórki dzięki występującym na ich powierzchni receptorom transferyny. Występują co najmniej dwa typy tych receptorów. Receptor transferyny 1 (TfR1) jest wytwarzany przez wszystkie komórki z wyjątkiem dojrzałych erytrocytów. Jego homologiem jest receptor transferyny 2 (TfR2), powstający głównie w hepatocytach [54]. Transport żelaza do wnętrza komórek w obydwu przypadkach odbywa się za pośrednictwem endocytozy [103]. Kompleks Tf-Fe łączy się z TfR i ulega internalizacji. W powstającym endosomie, pod wpływem obniżenia pH, żelazo odłącza się od transferyny i za pośrednictwem DMT1 przechodzi przez błonę endosomu do cytoplazmy. Komórkowe żelazo może być wykorzystane do wytwarzania hemu (w prekursorach erytrocytów) i innych zawierających żelazo białek lub magazynowane w postaci ferrytyny i hemosyderyny [22].
Do prawidłowego pobierania żelaza za pośrednictwem receptora transferyny niezbędne jest białko HFE. Właśnie mutacje w genie HFE, będące powodem syntezy zdefektowanego białka, sprzyjają gromadzeniu nadmiernej ilości żelaza w organizmie i są przez to główną przyczyną klasycznej, dziedzicznej hemochromatozy. HFE ulega ekspresji we wszystkich tkankach z wyjątkiem mózgu. W największej ilości powstaje w wątrobie i jelicie cienkim, co sugeruje udział zarówno w absorpcji, jak i magazynowaniu żelaza [31]. Białko HFE wykazuje homologię z cząsteczkami klasy I głównego układu zgodności tkankowej (MHC). Składa się z trzech domen zewnątrzkomórkowych (a
1, a
2 i a
3), domeny wewnątrzbłonowej i krótkiej części cytoplazmatycznej [30]. Z udziałem domeny a
1 łączy się z TfR1 [7]. Fragment a
3 wiąże się natomiast z b
2-mikroglobuliną (b
2M), co zapewnia właściwą orientację białka w błonie komórkowej [30]. Eksperymenty in vitro nie zdołały udowodnić możliwości wiązania HFE z TfR2 [111]. Udało się jednak zaobserwować interakcje tych białek in vivo [38]. Nieznany jest jednak mechanizm tego współdziałania. Natomiast TfR1 wymaga białka HFE do prawidłowego funkcjonowania. Brak HFE prowadzi do ograniczenia transportu żelaza do wnętrza komórek za pomocą tego receptora [104]. Aby mogło dojść do interakcji HFE z receptorem transferyny, niezbędna jest obecność b
2-mikroglobuliny. Jest to związane z ułatwieniem prezentacji białka HFE na powierzchni komórki [106].
W organizmie człowieka za kontrolowanie i uzupełnianie zawartości żelaza są odpowiedzialne komórki nabłonkowe dwunastnicy. Na podstawie występowania dużej ilości kompleksów HFE-b
2M-TfR1 na powierzchni komórek wyściełających krypty dwunastnicy zasugerowano mechanizm tego procesu [104]. Połączone z HFE receptory transferyny 1 wchłaniają do wnętrza komórek krypt znajdujące się w osoczu żelazo, co wpływa na osłabienie lub wzmocnienie ekspresji białek odpowiedzialnych za absorpcję i transport żelaza. W ten sposób ustalony zostaje pewien poziom zapotrzebowania na ten pierwiastek. Komórki krypt mają charakter multipotencjalnych komórek prekursorowych, z których część migruje do nabłonka kosmków jelitowych, przekształcając się w enterocyty [23,44] i staje się odpowiedzialna za pobranie zaprogramowanej ilości żelaza z przewodu pokarmowego. Na to jak dużo żelaza zostanie wchłonięte wpływają także substancje regulatorowe, sygnalizujące poziom tego pierwiastka w magazynach tkankowych oraz zapotrzebowanie na żelazo podczas erytropoezy. Jako mediatora odpowiadającego za zmiany ilości zmagazynowanego żelaza zaproponowano wytwarzaną w wątrobie hepcydynę [78]. W toku badań białko to uznano za główny czynnik regulujący homeostazę żelaza całego organizmu. Pierwotnie hepcydynie (liver expressed antimicrobial peptide-1 – LEAP-1), wyizolowanej z krwi [58] i z moczu [85], przypisano właściwości bakteriobójcze i antygrzybicze. Jednak prace Nicolasa i wsp. [78,79] wykazały duży wpływ tego białka zarówno na absorpcję żelaza w jelicie cienkim, jak i na – odpowiedzialny za recyrkulację tego metalu w organizmie – układ siateczkowo- śródbłonkowych makrofagów. Okazało się, że hepcydyna hamuje wchłanianie żelaza przez enterocyty oraz ogranicza uwalnianie żelaza z makrofagów. Dzieje się tak dzięki zdolności hepcydyny do blokowania ferroportyny, która jest jedynym znanym transporterem usuwającym żelazo z komórek [76]. Na zwiększenie ekspresji genu hepcydyny (HAMP) wpływ ma zarówno wzrost ilości żelaza w płynach ustrojowych, jak i wywołany infekcją bakteryjną lub wirusową, stan zapalny [89]. Nadmierne wytwarzanie hepcydyny może być jednak w każdym z tych przypadków zahamowane za pomocą mediatorów sygnalizujących wzmożoną erytropoezę [80]. Nieznane są dotąd czynniki regulatorowe wszystkich tych oddziaływań. Proponowano makrofagi jako komórki wysyłające tego typu sygnały [77], nowsze wyniki wskazują na hepatocyty [65].
Badania nad hepcydyną doprowadziły do zrewidowania hipotezy o regulacji absorpcji żelaza jedynie poprzez znajdujące się w komórkach krypt białko HFE. Stwierdzono, że hepcydyna wpływa na ten proces wiążąc i inaktywując ferroportynę w dojrzałych enterocytach kosmków jelitowych [33]. Nadal jednak widoczne są zależności między hepcydyną a białkiem HFE. Za pomocą nieznanych mechanizmów, nieprawidłowe białko HFE (np. zmutowane, jak u części pacjentów z dziedziczną hemochromatozą) powoduje osłabienie ekspresji genu hepcydyny (HAMP), co prowadzi do nadmiernego wchłaniania żelaza z pokarmu [16]. Na modelu mysim udało się zahamować ten proces poprzez konstytutywną ekspresję genu HAMP [81]. Potwierdzeniem ważnej roli hepcydyny w homeostazie żelaza było wykrycie mutacji w genie HAMP u osób cierpiących na niezwiązany z HFE typ hemochromatozy [95].
DZIEDZICZNA HEMOCHROMATOZA ZWIĄZANA Z POLIMORFIZMEM GENU HFE
Pierwsze przypuszczenia o tym, że hemochromatoza jest chorobą dziedziczną pojawiły się już w 1935 roku [101]. Ponad 40 lat później Simon i wsp. [102] określili lokalizację genu odpowiedzialnego za tę chorobę. Uważano wtedy, że HH wywołuje prosta mutacja umiejscowiona na krótkim ramieniu chromosomu 6. Dopiero w 1996 r. Feder i wsp. [31] zidentyfikowali gen hemochromatozy – HFE. Podczas wstępnej analizy stwierdzono obecność dwóch recesywnych mutacji zmiany sensu (missense) w badanym genie. U znacznej większości chorych na HH (ponad 80%) wykryto w obu allelach mutację powodującą zamianę cysteiny na tyrozynę – C282Y (Cys282Tyr). Natomiast około 4% pacjentów okazało się mieszanymi heterozygotami dla mutacji H63D (His63Asp) w połączeniu z heterozygotą C282Y. Kolejne badania potwierdziły te wyniki i przypisały osobom pochodzenia celtyckiego najczęstsze występowania polimorfizmu genu HFE [41]. Na podstawie analizy struktury białka HFE stwierdzono, że mutacja C282Y powoduje zmiany w konformacji domeny a
3, uniemożliwiając połączenie z b
2M. W wyniku tego HFE nie jest transportowane do powierzchni komórek, lecz pozostaje w retikulum endoplazmatycznym, gdzie ulega szybkiej degradacji. Mutacja H63D wpływa natomiast na strukturę domeny a
1. Jednak nie działa negatywnie na zdolność łączenia z TfR1 i prezentację komórkową [107]. Dlatego, jeżeli występuje samodzielnie zwykle nie powoduje choroby.
U osób cierpiących na HH, oprócz dwóch najczęściej opisywanych mutacji w genie HFE, obserwuje się także inne (tab. 1). Większość z nich wykrywana jest w pojedynczych przypadkach (w obrębie jednej rodziny) i jest współdziedziczona, podobnie jak H63D, z mutacją C282Y. Najbardziej znaczącą spośród nich jest mutacja S65C (Ser65Cys). Częstość występowania tego polimorfizmu określa się na 2–5% w populacji kaukaskiej [18]. S65C nie wpływa znacząco na zmianę struktury białka HFE i prowadzi do niewielkiego przeładowania organizmu żelazem, jeśli jest dziedziczona jako mieszana heterozygota z mutacją C282Y [75].
Tabela 1. Klasyfikacja odmian dziedzicznej hemochromatozy i wykaz zidentyfikowanych mutacji
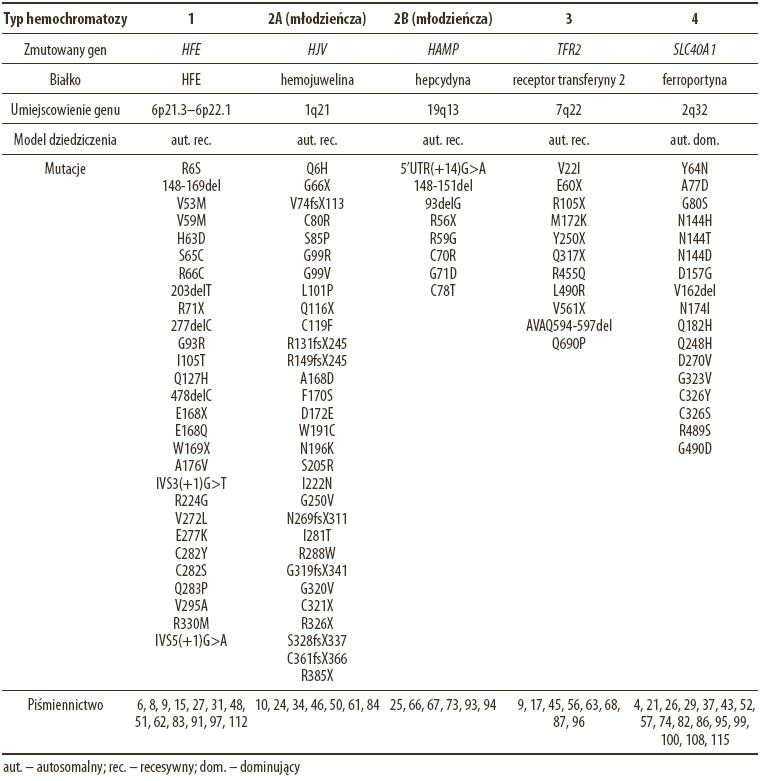
Należy również wspomnieć o dwóch nonsensownych mutacjach: E168X i W169X. Wśród mieszkańców północnych Włoch, chorych na hemochromatozę, występowały one w połączeniu z C282Y częściej niż H63D [90]. Pozostałe mutacje były opisywane jedynie w odosobnionych przypadkach [91]. Część z nich (np.: R71X, 478delC, IVS3(+1) G>T, IVS5(+1) G>A) znacząco wpływa na konstrukcję przestrzenną HFE powodując całkowitą utratę jego funkcji. Inne modyfikują bardziej (R66S, C282S, Q283P) lub mniej (R6S, G93R, R224G, R330M) strukturę tego białka.
Hemochromatoza wywołana mutacjami w genie HFE określona została jako typ 1 tej choroby. Chorują na nią głównie mężczyźni pochodzący z północnej Europy. Objawy ujawniają się zazwyczaj po 40. roku życia. Jest to najbardziej rozpowszechniona odmiana HH; dziedziczona jest autosomalnie w sposób recesywny. Charakteryzuje się nadmierną absorpcją żelaza z przewodu pokarmowego, co prowadzi do progresywnej akumulacji tego metalu w komórkach miąższowych wątroby, trzustki, serca i innych organów. Najwcześniejszym objawem jest wzrost wysycenia transferyny i stężenia ferrytyny we krwi. W zaawansowanym stadium choroby rozpoznawana jest wielonarządowa i nieodwracalna patologia (marskość wątroby, rak wątroby, cukrzyca, impotencja, kardiomiopatia, uogólniona artropatia) [5].
HEMOCHROMATOZA MŁODZIEŃCZA
Po odkryciu genu HFE zaczęto analizować genotypy pacjentów chorych na dziedziczną hemochromatozę. Okazało się, że wielu z nich ma mutacje w innych genach wpływających na metabolizm żelaza. Obserwowano także różnice w klinicznym obrazie choroby u tych osób. To wszystko doprowadziło do zidentyfikowania nowych postaci HH. Jedną z nich jest hemochromatoza młodzieńcza (juvenile hemochromatosis – JH) nazwana hemochromatozą typu 2; dziedziczona jest w sposób autosomalny, recesywny. Do jej rozwoju prowadzą mutacje w genie hepcydyny (HAMP) [95] lub hemojuweliny (HJV) [84]. Pierwsze oznaki choroby pojawiają się zazwyczaj przed 30. rokiem życia. Dotyka ona w równym stopniu obie płcie, a przebieg procesu gromadzenia żelaza w tkankach jest bardzo podobny do obserwowanego w typie 1 HH, jednak zachodzi dużo szybciej. Charakterystycznymi objawami są hipogonadyzm i kardiomiopatia. Nieleczona może doprowadzić do przedwczesnej śmierci spowodowanej niewydolnością serca.
Bardziej złośliwy przebieg młodzieńczej hemochromatozy jest wywołany mutacjami genu HAMP (typ 2B
). Czynna hepcydyna kodowana jest całkowicie przez trzeci ekson tego genu. Większość obserwowanych mutacji powoduje zmianę ramki odczytu właśnie w tym fragmencie [95]. Pierwsza z nich jest delecją guaniny w pozycji 93 (93delG). Jej następstwem jest powstanie wydłużonej o 95 aminokwasy prohepcydyny, której aktywna część pozbawiona jest swojej funkcji. Kolejna mutacja, zastąpienie cysteiny tyminą w pozycji 166, skutkuje przedwczesną terminacją (R56X) i zupełnym brakiem dojrzałej hepcydyny. Inne mutacje: C70R [66] i C78T [25] niszczą mostki dwusiarczkowe, będące podstawą przestrzennej struktury produktu genu HAMP. Nieprawidłowa hepcydyna przestaje ograniczać napływ jonów żelaza do organizmu za pośrednictwem ferroportyny, co wpływa na znaczne zwiększenie ilości tego metalu w tkankach.
Mutacja w genie HJV (HFE2) jest odpowiedzialna za łagodniejszą postać JH (typ 2A). Funkcja hemojuweliny nie została jeszcze dokładnie opisana, ale uważa się, że bierze ona udział w tym samym szlaku metabolicznym co hepcydyna i powoduje obniżenie jej ekspresji [84]. Hemojuvelina jest białkiem transmembranowym powstającym w wątrobie, sercu i mięśniach szkieletowych. Obecnie znanych jest ponad 30 chorobotwórczych mutacji w genie HJV, z których większość wykryto jedynie w pojedynczych przypadkach (tab. 1). Najczęściej powodują one przedwczesną terminację lub modyfikują konserwowane aminokwasy. Najbardziej rozpowszechnioną jest substytucja G320V, rozpoznana u pacjentów pochodzących z Europy, Ameryki i Azji [60]. Papanikolaou i wsp. znaleźli tę mutację u 80% badanych już podczas wstępnej analizy genu hemojuweliny [84].
INNE POSTACI DZIEDZICZNEJ HEMOCHROMATOZY
Duży odsetek chorych na HH, u których nie wykrywa się mutacji w genie HFE obserwuje się we Włoszech (ponad 35%) [19]. Analizując grupę pacjentów z tego kraju, Camaschella i wsp. [17] zidentyfikowali gen TFR2 odpowiedzialny za nową odmianę hemochromatozy – typ 3. Mimo oznaczenia młodzieńczej hemochromatozy jako typ 2, to gen receptora transferyny 2 udało się wykryć jako pierwszy po HFE, wcześniej niż geny HAMP czy HJV. Fenotyp kliniczny pacjentów cierpiących na hemochromatozę wywołaną mutacjami TFR2 jest bardzo zbliżony do tego obserwowanego w typie 1 HH, jednak objawy pojawiają się nieco wcześniej [36]. Pierwszą opisaną mutacją była transwersja cytozyny na guaninę w pozycji 750 kodującego mRNA (Y250X), wykryta u dwóch niespokrewnionych sycylijskich rodzin [17]. Kolejne mutacje (tab. 1), tak jak pierwsza, miały charakter autosomalny, recesywny i w większości odkryto je wśród osób pochodzenia włoskiego. Warto podkreślić, że zidentyfikowano trzy mutacje w genie TFR2 (L490R, V561X i AVAQ 594-597) u pacjentów japońskich [42,57]. Japończycy w odróżnieniu od przedstawicieli populacji kaukaskiej bardzo rzadko chorują na hemochromatozę.
Rola receptora transferyny 2 w regulowaniu homeostazy żelaza nie została jeszcze określona. Fleming i wsp. proponują model, w którym TfR2 spełnia funkcję wątrobowego sensora wykrywającego ilość żelaza w płynach ustrojowych [32]. Według nich receptor transferyny 2 jest odpowiedzialny za pobieranie żelaza przez hepatocyty. Intensywność tego procesu zależy od stopnia wysycenia transferyny we krwi. To, ile żelaza dostało się do hepatocytów wpływa natomiast na ilość syntetyzowanej hepcydyny. Nieprawidłowe białko TfR2 nie zwiększa więc ekspresji genu HAMP w wypadku nadmiaru żelaza w organizmie, przez co absorpcja tego metalu z pożywienia nie zostaje zahamowana. Potwierdza to praca Wallace’a i wsp., którzy stwierdzili brak wzmożonej syntezy hepcydyny w odpowiedzi na przeładowanie żelazem u myszy pozbawionych genu TFR2 [109].
Badania genotypów mieszkańców Wysp Salomona doprowadziły do identyfikacji kolejnego, 4 typu dziedzicznej hemochromatozy [4]. Ta odmiana choroby dotyka głównie dorosłych, dziedziczona jest w sposób autosomalny dominujący a wywołują ją mutacje w genie kodującym ferroportynę (SLC40A1, nazywany poprzednio SLC11A3). Ze względu na specyficzny obraz kliniczny wielu autorów uznaje ją za odrębną jednostkę chorobową (ferroportin disease) [86,20]. Analizy kolejnych grup pacjentów doprowadziły do wykrycia mutacji w genie SLC40A1 wśród mieszkańców Ameryki Północnej, Afryki, Europy i Azji (tab. 1). Wartą wyróżnienia jest tutaj delecja waliny w pozycji 162 (V162del), którą stwierdzono u chorych z Australii, Grecji, Wielkiej Brytanii i Włoch [21,26,94,108]. Częste występowanie wśród Afrykańczyków i Amerykanów pochodzenia afrykańskiego obserwuje się dla substytucji Q248H [37]. Pozostałe mutacje mają zazwyczaj charakter jednostkowych przypadków.
Efekty kliniczne mutacji w genie SLC40A1 nie zawsze są jednakowe. Wynika to przede wszystkim ze złożoności oddziaływań ferroportyny z innymi białkami i jej skomplikowanej budowy, przez co miejsce wystąpienia mutacji ma duży wpływ na obraz choroby. Za objawy charakterystyczne dla HH typu 4 uznaje się gromadzenie żelaza w wątrobie w makrofagach (komórkach Kupffera), a nie hepatocytach oraz zawyżone stężenie ferrytyny w surowicy przy prawidłowym lub nieco podwyższonym wysyceniu transferyny [86]. W przeciwieństwie do innych typów hemochromatozy, w przypadku HH typu 4 leczenie upustami krwi nie może być stosowane, ponieważ prowadzi do anemii [88]. Anemię rozpoznaje się często u kobiet miesiączkujących, nosicielek mutacji w genie SLC40A1.
Ferroportyna jest transbłonowym białkiem spełniającym funkcje transportera usuwającego żelazo z komórek. Ulega ona ekspresji w komórkach, które odgrywają dużą rolę w metabolizmie tego metalu: głównie w komórkach Kupffera i enterocytach kosmków jelitowych, ale także w hepatocytach, makrofagach układu siateczkowo-śródbłonkowego i syncycjotrofoblaście łożyska [1,72]. Przypuszcza się, że struktura przestrzenna ferroportyny składa się z 9 lub 10 transmembranowych helis [26,28]. Mimo to, że chorobotwórcze mutacje w genie SLC40A1 obejmują całe białko, większość z nich (12 z 17) skupia się między pierwszą a czwartą transmembranową domeną. Ta część ferroportyny może być odpowiedzialna za transport żelaza, albo jest miejscem interakcji z hefajstyną lub hepcydyną, które wpływają odpowiednio synergistycznie [106] i antagonistycznie [76] na eksport żelaza z komórki. Wpływ rodzaju mutacji na prezentację kliniczną choroby próbowali wyjaśnić Schimanski i wsp. [29,99]. Wykazali oni, że ferroportyna z mutacjami A77D, V162del i G490D trudniej osiąga powierzchnię komórki przez co transport żelaza jest osłabiony. Zmiany Y64N, N144D, N144H i C326Y powodują natomiast odporność ferroportyny na inhibicję za pośrednictwem hepcydyny. W przypadku pierwszej grupy mutacji produkt genu SLC40A1 pozbawiony jest swojej funkcji. Następuje retencja żelaza w makrofagach (odpowiedzialnych za odzyskiwanie żelaza ze starych erytrocytów), co prowadzi do akumulacji tego metalu w tkankach i objawia się wzrostem stężenia ferrytyny w osoczu. Zmniejszenie ilości żelaza w układzie krążenia wpływa natomiast na obniżony poziom wysycenia transferyny oraz na system hematopoetyczny, który zaczyna indukować wchłanianie żelaza z pożywienia [74]. Druga grupa mutacji jest związana natomiast ze wzrostem aktywności ferroportyny. Brak wrażliwości na hepcydynę powoduje nadmierną absorpcję żelaza z przewodu pokarmowego i zmniejszenie ilości tego metalu w makrofagach. Rośnie ilość żelaza w osoczu. W tym przypadku obraz choroby charakteryzuje się wzrostem wysycenia transferyny i zbliżony jest do tego obserwowanego w pozostałych odmianach dziedzicznej hemochromatozy [100].
Choroba wywołana mutacją podjednostki ciężkiej ferrytyny (H-ferrytyny) uznawana była przez niektórych autorów za typ 5 HH [2,12]. Kontrola ekspresji ferrytyny odbywa się poprzez substancje sygnalizujące ilość żelaza (iron responsive element – IRE) znajdujące się na końcu 5’ mRNA tego białka. Właśnie we fragmencie IRE H-ferrytyny wykryto dominującą mutację A49U, która powoduje nadmierne gromadzenie żelaza w organizmie [53]. Schorzeniu temu towarzyszy wzrost wysycenia transferyny i duże stężenie ferrytyny.
DWUGENOWY MODEL DZIEDZICZNEJ HEMOCHROMATOZY
Na przebieg procesu spichrzania żelaza u chorych na HH wpływa wiele modyfikujących czynników genetycznych i środowiskowych. Ilości nagromadzonego w ustroju żelaza u osób z mutacją C282Y w genie HFE różnią się nawet dziesięciokrotnie [13]. Uwzględniając to, kilka grup badawczych zaczęło analizować genotypy pacjentów, będących heterozygotycznymi nosicielami mutacji C282Y [9,49,73]. U wielu z nich potwierdzono obecność dodatkowych mutacji w genie HAMP, co zasugerowało możliwość dwugenowego modelu dziedziczenia hemochromatozy. Rodzaj mutacji w genie hepcydyny miał wpływ na obraz choroby [73]. Delecja czterech nukleotydów na końcu 3’ eksonu 2 (Met50del IVS2+1 (–G)), skutkująca brakiem aktywnego białka, powodowała młodzieńczą postać hemochromatozy o złym rokowaniu. Mutacja G71D w połączeniu z heterozygotyczną mutacją C282Y w genie HFE wywoływała klasyczny 1 typ HH. Inne badania wykazały, że przyczyną młodzieńczej hemochromatozy może być również synergistyczny efekt mutacji w genach HFE i TFR2 [87]. Te wszystkie spostrzeżenia tłumaczą heterogeniczność fenotypową wśród osób chorujących na HH. Ich interpretacja nie jest jednak jednoznaczna, głównie ze względu na bardzo małą liczbę zbadanych przypadków.
PODSUMOWANIE
HH jest złożoną chorobą genetyczną. Powiązanie genów HFE, HJV, HAMP, TFR2 i SLC40A1 z patologicznym procesem gromadzenia żelaza pozwala przypuszczać, że wszystkie one biorą udział w jednym szlaku procesowym. Uwzględniając nadrzędną rolę hepcydyny, produktom pozostałych genów można przypisać rolę regulatorów jej aktywności i ekspresji. Lepsze poznanie tych zależności wymaga identyfikacji kolejnych białek i mechanizmów rządzących homeostazą żelaza. Mogłoby to znacznie ułatwić diagnozowanie i leczenie dziedzicznej hemochromatozy.
PIŚMIENNICTWO
[1] Abboud S., Haile D.J.: A novel mammalian ironregulated protein involved in intracellular iron metabolism. J. Biol. Chem., 2000; 275: 19906-19912
[PubMed] [Full Text HTML] [Full Text PDF]
[2] Anderson G.J., Powell L.W.: HFE and non-HFE hemochromatosis. Int. J. Hematol., 2002; 76: 203-207
[PubMed]
[3] Andrews N.C.: Disorders of iron metabolism. N. Engl. J. Med., 1999; 341: 1986-1995
[PubMed]
[4] Arden K.E., Wallace D.F., Dixon J.L., Summerville L., Searle J.W., Anderson G.J., Ramm G.A., Powell L.W., Subramaniam V.N.: A novel mutation in ferroportin1 is associated with haemochromatosis in a Solomon Islands patient. Gut, 2003; 52: 1215-1217
[PubMed] [Full Text HTML] [Full Text PDF]
[5] Bacon B.R.: Hemochromatosis: diagnosis and management. Gastroenterology, 2001; 120: 718-725
[PubMed]
[6] Barton J.C., Sawada-Hirai R., Rothenberg B.E., Acton R.T.: Two novel missense mutations in the HFE gene (I105T and G93R) and identification of the S65C mutation in Alabama hemochromatosis probands. Blood. Cell. Mol. Dis., 1999; 25: 147-155
[PubMed]
[7] Bennett M.J., Lebron J.A., Bjorkman P.J.: Crystal structure of the haemochromatosis protein HFE complexed with transferrin receptor. Nature, 2000; 403: 46-53
[PubMed]
[8] Beutler E., Griffin M.J., Gelbart T., West C.: A previously undescribed nonsense mutation of the HFE gene. Clin. Genet., 2002; 61: 40-42
[PubMed]
[9] Biasiotto G., Belloli S., Ruggeri G., Zanella I., Gerardi G., Corrado M., Gobbi E., Albertini A., Arosio P.: Identification of new mutations of the HFE, hepcidin, and transferrin receptor 2 genes by denaturing HPLC analysis of individuals with biochemical indications of iron overload. Clin. Chem., 2003; 49: 1981-1988
[PubMed] [Full Text HTML] [Full Text PDF]
[10] Biasiotto G., Roetto A., Daraio F., Polotti A., Gerardi G.M., Girelli D., Cremonesi L., Arosio P., Camaschella C.: Identification of new mutations of hepcidin and hemojuvelin in patients with HFE C282Y allele. Blood Cells Mol. Dis., 2004; 33: 338-343
[PubMed]
[11] Boldt D.H.: New perspectives on iron: an introduction. Am. J. Med. Sci., 1999; 318: 207-212
[PubMed]
[12] Bomford A.: Genetics of haemochromatosis. Lancet, 2002; 360: 1673-1681
[PubMed]
[13] Bothwell T.H., McPhail A.P.: Hereditary haemochromatosis: etiologic, pathologic and clinical aspects. Semin. Hematol., 1998; 35: 55-71
[PubMed]
[14] Bottomley S.S., May B.K., Cox T.C., Cotter P.D., Bishop D.F.: Molecular defects of erythroid 5-aminolevulinate synthase in X-linked sideroblastic anemia. J. Bioenerg. Biomembr., 1995; 27: 161-168
[PubMed]
[15] Bradbury R., Fagan E., Payne S.J.: Two novel polymorphisms (E277K and V212V) in the haemochromatosis gene HFE. Hum. Mutat., 2000; 15: 120
[PubMed]
[16] Bridle K.R., Frazer D.M., Wilkins S.J., Dixon J.L., Purdie D.M., Crawford D.H., Subramaniam V.N., Powell L.W., Anderson G.J., Ramm G.A.: Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homeostasis. Lancet, 2003; 361: 669-673
[PubMed]
[17] Camaschella C., Roetto A., Cali A., De Gobbi M., Garozzo G., Carella M., Majorano N., Totaro A., Gasparini P.: The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet., 2000; 25: 14-15
[PubMed] [Full Text HTML] [Full Text PDF]
[18] Camaschella C., Roetto A., De Gobbi M.: Genetic haemochromatosis: genes and mutations associated with iron loading. Best. Pract. Res. Clin. Haematol., 2002; 15: 261-276
[PubMed]
[19] Carella M., D’Ambrosio L., Totaro A., Grifa A., Valentino M.A., Piperno A., Girelli D., Roetto A., Franco B., Gasparini P., Camaschella C.: Mutation analysis of the HLA-H gene in Italian hemochromatosis patients. Am. J. Hum. Genet., 1997; 60: 828-832
[PubMed]
[20] Cazzola M.: Genetic disorders of iron overload and the novel 'ferroportin disease’. Haematologica, 2003; 88: 721-724
[PubMed]
[21] Cazzola M., Cremonesi L., Papaioannou M., Soriani N., Kioumi A., Charalambidou A., Paroni R., Romtsou K., Levi S., Ferrari M., Arosio P., Christakis J.: Genetic hyperferritinaemia and reticuloendothelial iron overload associated with a three base pair deletion in the coding region of the ferroportin gene (SLC11A3). Br. J. Haematol., 2002; 119: 539-546
[PubMed]
[22] Conrad M.E., Umbreit J.N., Moore E.G.: Iron absorption and transport. Am. J. Med. Sci., 1999; 318: 213-229
[PubMed]
[23] Daniele B., D’Agostino L.: Proliferation and differentiation of the small intestinal epithelium: from petri dish to bedside. Ital. J. Gastroenterol., 1994; 26: 459-470
[PubMed]
[24] Daraio F., Ryan E., Gleeson F., Roetto A., Crowe J., Camaschella C.: Juvenile hemochromatosis due to G320V/Q116X compound heterozygosity of hemojuvelin in an Irish patient. Blood Cells Mol. Dis., 2005; 35: 174-176
[PubMed]
[25] Delatycki M.B., Allen K.J., Gow P., MacFarlane J., Radomski C., Thompson J., Hayden M.R., Goldberg Y.P., Samuels M.E.: A homozygous HAMP mutation in a multiply consanguineous family with pseudodominant juvenile hemochromatosis. Clin. Genet., 2004; 65: 378-383
[PubMed]
[26] Devalia V., Carter K., Walker A.P., Perkins S.J., Worwood M., May A., Dooley J.S.: Autosomal dominant reticuloendothelial iron overload associated with a 3-base pair deletion in the ferroportin 1 gene (SLC11A3). Blood, 2002; 100: 695-697
[PubMed] [Full Text HTML] [Full Text PDF]
[27] de Villiers J.N., Hillermann R., Loubser L., Kotze M.J.: Spectrum of mutations in the HFE gene implicated in haemochromatosis and porphyria. Hum. Mol. Genet., 1999; 8: 1517-1522
[PubMed] [Full Text HTML] [Full Text PDF]
[28] Donovan A., Brownlie A., Zhou Y., Shepard J., Pratt S.J., Moynihan J., Paw B.H., Drejer A., Barut B., Zapata A., Law T.C., Brugnara C., Lux S.E., Pinkus G.S., Pinkus J.L., Kingsley P.D., Palis J., Fleming M.D., Andrews N.C., Zon L.I.: Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature, 2000; 403: 776-781
[PubMed]
[29] Drakesmith H., Schimanski L.M., Ormerod E., Merryweather-Clarke A.T., Viprakasit V., Edwards J.P., Sweetland E., Bastin J.M., Cowley D., Chinthammitr Y., Robson K.J., Townsend A.R.: Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood, 2005; 106: 1092-1097
[PubMed]
[30] Ehrlich R., Lemonnier F.A.: HFE: a novel nonclassical class I molecule that is involved in iron metabolism. Immunity, 2000; 13: 585-588
[PubMed]
[31] Feder J.N., Gnirke A., Thomas W., Tsuchihashi Z., Ruddy D.A., Basava A., Dormishian F., Domingo R.Jr, Ellis M.C., Fullan A., Hinton L.M., Jones N.L., Kimmel B.E., Kronmal G.S., Lauer P., Lee V.K., Loeb D.B., Mapa F.A., McClelland E., Meyer N.C., Mintier G.A., Moeller N., Moore T., Morikang E., Prass C.E., Quintana L., Starnes S.M., Schatzman R.C., Brunke K.J., Drayna D.T., Risch N.J., Bacon B.R., Wolff R.K.: A novel MHC class I-like gene is mutated in patients with hereditary hemochromatosis. Nat. Genet., 1996; 13: 399-408
[PubMed]
[32] Fleming R.E., Ahmann J.R., Migas M.C., Waheed A., Koeffler H.P., Kawabata H., Britton R.S., Bacon B.R., Sly W.S.: Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc. Natl. Acad. Sci. USA, 2002; 99: 10653-10658
[PubMed] [Full Text HTML] [Full Text PDF]
[33] Ganz T.: Hepcidin — a regulator of intestinal iron absorption and iron recycling by macrophages. Best. Pract. Res. Clin. Haematol., 2005; 18: 171-182
[PubMed]
[34] Gehrke S.G., Pietrangelo A., Kascak M., Braner A., Eisold M., Kulaksiz H., Herrmann T., Hebling U., Bents K., Gugler R., Stremmel W.: HJV gene mutations in European patients with juvenile hemochromatosis. Clin. Genet., 2005; 67: 425-428
[PubMed]
[35] Gerlach M., Ben-Shachar D., Riederer P., Youdim M.B.: Altered brain metabolism of iron as a cause of neurodegenerative diseases? J. Neurochem., 1994; 63: 793-807
[PubMed] [Full Text PDF]
[36] Girelli D., Bozzini C., Roetto A., Alberti F., Daraio F., Colombari R., Olivieri O., Corrocher R., Camaschella C.: Clinical and pathologic findings in hemochromatosis type 3 due to a novel mutation in transferrin receptor 2 gene. Gastroenterology, 2002; 122: 1295-1302
[PubMed]
[37] Gordeuk V.R., Caleffi A., Corradini E., Ferrara F., Jones R.A., Castro O., Onyekwere O., Kittles R., Pignatti E., Montosi G., Garuti C., Gangaidzo I.T., Gomo Z.A., Moyo V.M., Rouault T.A., MacPhail P., Pietrangelo A.: Iron overload in Africans and African-Americans and a common mutation in the SCL40A1 (ferroportin 1) gene. Blood Cells Mol. Dis., 2003; 31: 299-304
[PubMed]
[38] Griffiths W.J., Cox T.M.: Co-localization of the mammalian hemochromatosis gene product (HFE) and a newly identified transferrin receptor (TfR2) in intestinal tissue and cells. J. Histochem. Cytochem., 2003; 51: 613-624
[PubMed] [Full Text HTML] [Full Text PDF]
[39] Gunshin H., Mackenzie B., Berger U.V., Gunshin Y., Romero M.F., Boron W.F., Nussberger S., Gollan J.L., Hediger M.A.: Cloning and characterization of a mammalian protoncoupled metal-ion transporter. Nature, 1997; 388: 482-488
[PubMed]
[40] Halliwell B., Gutteridge J.M.: Biologically relevant metal ion-dependent hydroxyl radical generation. An update. FEBS Lett., 1992; 307: 108-112
[PubMed]
[41] Hanson E.H., Imperatore G., Burke W.: HFE gene and hereditary hemochromatosis: a HuGE review. Human Genome Epidemiology. Am. J. Epidemiol., 2001; 154: 193-206
[PubMed] [Full Text HTML] [Full Text PDF]
[42] Hattori A., Wakusawa S., Hayashi H., Harashima A., Sanae F., Kawanaka M., Yamada G., Yano M., Yoshioka K.: AVAQ 594-597 deletion of the TFR2 gene in a Japanese family with hemochromatosis. Hepatol. Res., 2003; 26: 154-156
[PubMed]
[43] Hetet G., Devaux I., Soufir N., Grandchamp B., Beaumont C.: Molecular analyses of patients with hyperferritinemia and normal serum iron values reveal both L ferritin IRE and 3 new ferroportin (slc11A3) mutations. Blood, 2003; 102: 1904-1910
[PubMed] [Full Text HTML] [Full Text PDF]
[44] Hocker M., Weidenmann B.: Molecular mechanisms of enteroendocrine differentiation. Ann. N. Y. Acad. Sci., 1998; 859: 160-174
[PubMed]
[45] Hofmann W.K., Tong X.J., Ajioka R.S., Kushner J.P., Koeffler H.P.: Mutation analysis of transferrin-receptor 2 in patients with atypical hemochromatosis. Blood, 2002; 100: 1099-1100
[PubMed] [Full Text HTML] [Full Text PDF]
[46] Huang F.W., Rubio-Aliaga I., Kushner J.P., Andrews N.C., Fleming M.D.: Identification of a novel mutation (C321X) in HJV. Blood, 2004; 104: 2176-2177
[PubMed] [Full Text HTML] [Full Text PDF]
[47] Huebers H.A., Csiba E., Huebers E., Finch C.A.: Competitive advantage of diferric transferrin in delivering iron to reticulocytes. Proc. Natl. Acad. Sci. USA, 1983; 80: 300-304
[PubMed] [Full Text HTML] [Full Text PDF]
[48] Imanishi H., Liu W., Cheng J., Ikeda N., Amuro Y., Hada T.: Idiopathic hemochromatosis with the mutation of Ala176Val heterozygous for HFE gene. Intern. Med., 2001; 40: 479-483
[PubMed]
[49] Jacolot S., Le Gac G., Scotet V., Quere I., Mura C., Ferec C.: HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype. Blood, 2004; 103: 2835-2840
[PubMed] [Full Text HTML] [Full Text PDF]
[50] Janosi A., Andrikovics H., Vas K., Bors A., Hubay M., Sapi Z., Tordai A.: Homozygosity for a novel nonsense mutation (G66X) of the HJV gene causes severe juvenile hemochromatosis with fatal cardiomyopathy. Blood, 2005; 105: 432
[PubMed] [Full Text HTML] [Full Text PDF]
[51] Jones D.C., Young N.T., Pigott C., Fuggle S.V., Barnardo M.C., Marshall S.E., Bunce M.: Comprehensive hereditary hemochromatosis genotyping. Tissue Antigens, 2002; 60: 481-488
[PubMed]
[52] Jouanolle A.M., Douabin-Gicquel V., Halimi C., Loreal O., Fergelot P., Delacour T., de Lajarte-Thirouard A.S., Turlin B., Le Gall J.Y., Cadet E., Rochette J., David V., Brissot P.: Novel mutation in ferroportin 1 gene is associated with autosomal dominant iron overload. J. Hepatol., 2003; 39: 286-289
[PubMed]
[53] Kato J., Fujikawa K., Kanda M., Fukuda N., Sasaki K., Takayama T., Kobune M., Takada K., Takimoto R., Hamada H., Ikeda T., Niitsu Y.: A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am. J. Hum. Genet., 2001; 69: 191-197
[PubMed] [Full Text HTML] [Full Text PDF]
[54] Kawabata H., Yang R., Hirama T., Vuong P.T., Kawano S., Gombart A.F., Koeffler H.P.: Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem., 1999; 274: 20826-20832
[PubMed] [Full Text HTML] [Full Text PDF]
[55] Kelly A.L., Lunt P.W., Rodrigues F., Berry P.J., Flynn D.M., McKiernan P.J., Kelly D.A., Mieli-Vergani G., Cox T.M.: Classification and genetic features of neonatal haemochromatosis: a study of 27 affected pedigrees and molecular analysis of genes implicated in iron metabolism. J. Med. Genet., 2001; 38: 599-610
[PubMed] [Full Text HTML] [Full Text PDF]
[56] Koyama C., Wakusawa S., Hayashi H., Suzuki R., Yano M., Yoshioka K., Kozuru M., Takayamam Y., Okada T., Mabuchi H.: Two novel mutations, L490R and V561X, of the transferrin receptor 2 gene in Japanese patients with hemochromatosis. Haematologica, 2005; 90: 302-307
[PubMed]
[57] Koyama C., Wakusawa S., Hayashi H., Ueno T., Suzuki R., Yano M., Saito H., Okazaki T.: A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern. Med., 2005; 44: 990-993
[PubMed] [Full Text PDF]
[58] Krause A., Neitz S., Magert H.J., Schulz A., Forssmann W.G., Schulz-Knappe P., Adermann K.: LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett., 2000; 480: 147-150
[PubMed]
[59] Lanzara C., Roetto A., Daraio F., Rivard S.R., Ficarella R., Simard H., Cox T.M., Cazzola M., Piperno A., Gimenez-Roqueplo A-P., Grammatico P., Volinia S., Gasparini P., Camaschella C.: The spectrum of hemojuvelin gene mutation in 1q-linked juvenile hemochromatosis. Blood, 2004; 103: 4317-4321
[PubMed] [Full Text HTML] [Full Text PDF]
[60] Lee P.L., Barton J.C., Brandhagen D., Beutler E.: Hemojuvelin (HJV) mutations in persons of European, African-American and Asian ancestry with adult onset haemochromatosis. Br. J. Haematol., 2004; 127: 224-229
[PubMed]
[61] Lee P.L., Beutler E., Rao S.V., Barton J.C.: Genetic abnormalities and juvenile hemochromatosis: mutations of the HJV gene encoding hemojuvelin. Blood, 2004; 103: 4669-4671
[PubMed] [Full Text HTML] [Full Text PDF]
[62] Le Gac G., Dupradeau F.Y., Mura C., Jacalot S., Scotet V., Esnault G., Mercier A.Y., Rochette J., Ferec C.: Phenotypic expression of the C282Y/Q283P compound heterozygosity in HFE and molecular modeling of the Q283P mutation effect. Blood. Cell. Mol. Dis., 2003; 30: 231-237
[PubMed]
[63] Le Gac G., Mons F., Jacolot S., Scotet V., Ferec C., Frebourg T.: Early onset hereditary hemochromatosis resulting from a novel TFR2 gene nonsense mutation (R105X) in two siblings of north French descent. Br. J. Haematol., 2004; 125: 674-678
[PubMed]
[64] Lieu P.T., Heiskala M., Peterson P.A., Yang Y.: The roles of iron in health and disease. Mol. Aspects. Med., 2001; 22: 1-87
[PubMed]
[65] Lou D.Q., Lesbordes J.C., Nicolas G., Viatte L., Bennoun M., Van Rooijen N., Kahn A., Renia L., Vaulont S.: Iron- and inflammation-induced hepcidin gene expression in mice is not mediated by Kupffer cells in vivo. Hepatology, 2005; 41: 1056-1064
[PubMed]
[66] Majore S., Binni F., Pennese A., De Santis A., Crisi A., Grammatico P.: HAMP gene mutation c.208T>C (p.C70R) identified in an Italian patient with severe hereditary hemochromatosis. Hum. Mutat., 2004; 23: 400
[PubMed]
[67] Matthes T., Aguilar-Martinez P., Pizzi-Bosman L., Darbellay R., Rubbia-Brandt L., Giostra E., Michel M., Ganz T., Beris P.: Severe hemochromatosis in a Portuguese family associated with a new mutation in the 5′-UTR of the HAMP gene. Blood, 2004; 104: 2181-2183
[PubMed] [Full Text HTML] [Full Text PDF]
[68] Mattman A., Huntsman D., Lockitch G., Langlois S., Buskard N., Ralston D., Butterfield Y., Rodrigues P., Jones S., Porto G., Marra M., De Sousa M., Vatcher G.: Transferrin receptor 2 (TfR2) and HFE mutational analysis in non-C282Y iron overload: identification of a novel TfR2 mutation. Blood, 2002; 100: 1075-1077
[PubMed] [Full Text HTML] [Full Text PDF]
[69] May B.K., Dogra S.C., Sadlon T.J., Bhasker C.R., Cox T.C., Bottomley S.S.: Molecular regulation of heme biosynthesis in higher vertebrates. Prog. Nucleic. Acid. Res. Mol. Biol., 1995; 51: 1-51
[PubMed]
[70] McCord J.M.: Iron, free radicals, and oxidative injury. Semin. Hematol., 1998; 35: 5-12
[PubMed]
[71] McKie A.T., Barrow D., Latunde-Dada G.O., Rolfs A., Sager G., Mudaly E., Mudaly M., Richardson C., Barlow D., Bomford A., Peters T.J., Raja K.B., Shirali S., Hediger M.A., Farzaneh F., Simpson R.J.: An iron-regulated ferric reductase associated with the absorption of dietary iron. Science, 2001; 291: 1755-1759
[PubMed] [Full Text HTML] [Full Text PDF]
[72] McKie A.T., Marciani P., Rolfs A., Brennan K., Wehr K., Barrow D., Miret S., Bomford A., Peters T.J., Farzaneh F., Hediger M.A., Hentze M.W., Simpson R.J.: A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell., 2000; 5: 299-309
[PubMed]
[73] Merryweather-Clarke A.T., Cadet E., Bomford A., Capron D., Viprakasit V., Miller A., McHugh P.J., Chapman R.W., Pointon J.J., Wimhurst V.L., Livesey K.J., Tanphaichitr V., Rochette J., Robson K.J.: Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum. Mol. Genet., 2003; 12: 2241-2247
[PubMed] [Full Text HTML] [Full Text PDF]
[74] Montosi G., Donovan A., Totaro A., Garuti C., Pignatti E., Cassanelli S., Trenor C.C., Gasparini P., Andrews N.C., Pietrangelo A.: Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J. Clin. Invest., 2001; 108: 619-623
[PubMed] [Full Text HTML] [Full Text PDF]
[75] Mura C., Raguenes O., Ferec C.: HFE mutations analysis in 711 hemochromatosis probands: evidence for S65C implication in mild form of hemochromatosis. Blood, 1999; 93: 2502-2505
[PubMed] [Full Text HTML] [Full Text PDF]
[76] Nemeth E., Tuttle M.S., Powelson J., Vaughn M.B., Donovan A., Ward D.M., Ganz T., Kaplan J.: Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science, 2004; 306: 2090-2093
[PubMed] [Full Text HTML] [Full Text PDF]
[77] Nemeth E., Valore E.V., Territo M., Schiller G., Lichtenstein A., Ganz T.: Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood, 2003; 101: 2461-2463
[PubMed] [Full Text HTML] [Full Text PDF]
[78] Nicolas G., Bennoun M., Devaux I., Beaumont C., Grandchamp B., Kahn A., Vaulont S.: Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA, 2001; 98: 8780-8785
[PubMed] [Full Text HTML] [Full Text PDF]
[79] Nicolas G., Bennoun M., Porteu A., Mativet S., Beaumont C., Grandchamp B., Sirito M., Sawadogo M., Kahn A., Vaulont S.: Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. USA, 2002; 99: 4596-4601
[PubMed] [Full Text HTML] [Full Text PDF]
[80] Nicolas G., Chauvet C., Viatte L., Danan J.L., Bigard X., Devaux I., Beaumont C., Kahn A., Vaulont S.: The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest., 2002; 110: 1037-1044
[PubMed] [Full Text HTML] [Full Text PDF]
[81] Nicolas G., Viatte L., Lou D.Q., Bennoun M., Beaumont C., Kahn A., Andrews N.C., Vaulont S.: Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat. Genet., 2003; 34: 97-101
[PubMed]
[82] Njajou O.T., Vaessen N., Joosse M., Berghuis B., van Dongen J.W., Breuning M.H., Snijders P.J., Rutten W.P., Sandkuijl L.A., Oostra B.A., van Duijn C.M., Heutink P.: A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat. Genet., 2001; 28: 213-214
[PubMed] [Full Text HTML] [Full Text PDF]
[83] Oberkanins C., Moritz A., de Villiers J.N., Kotze M.J., Kury F.: A reverse-hybridization assay for the rapid and simultaneous detection of nine HFE gene mutations. Genet. Test., 2000; 4: 121-124
[PubMed]
[84] Papanikolaou G., Samuels M.E., Ludwig E.H., MacDonald M.L., Franchini P.L., Dube M.P., Andres L., MacFarlane J., Sakellaropoulos N., Politou M., Nemeth E., Thompson J., Risler J.K., Zaborowska C., Babakaiff R., Radomski C.C., Pape T.D., Davidas O., Christakis J., Brissot P., Lockitch G., Ganz T., Hayden M.R., Goldberg Y.P.: Mutations in HFE2 cause iron overload in chromosome 1qlinked juvenile hemochromatosis. Nat. Genet., 2004; 36: 77-82
[PubMed]
[85] Park C.H., Valore E.V., Waring A.J., Ganz T.: Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem., 2001; 276: 7806-7810
[PubMed] [Full Text HTML] [Full Text PDF]
[86] Pietrangelo A.: The ferroportin disease. Blood. Cells. Mol. Dis., 2004; 32: 131-138
[PubMed]
[87] Pietrangelo A., Caleffi A., Henrion J., Ferrara F., Corradini E., Kulaksiz H., Stremmel W., Andreone P., Garuti C.: Juvenile hemochromatosis associated with pathogenic mutations of adult hemochromatosis genes. Gastroenterology, 2005; 128: 470-479
[PubMed]
[88] Pietrangelo A., Montosi G., Totaro A., Garuti C., Conte D., Cassanelli S., Fraquelli M., Sardini C., Vasta F., Gasparini P.: Hereditary hemochromatosis in adults without pathogenic mutations in the hemochromatosis gene. N. Engl. J. Med., 1999; 341: 725-732
[PubMed]
[89] Pigeon C., Ilyin G., Courselaud B., Leroyer P., Turlin B., Brissot P., Loreal O.: A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem., 2001; 276: 7811-7819
[PubMed] [Full Text HTML] [Full Text PDF]
[90] Piperno A., Arosio C., Fossati L., Vigano M., Trombini P., Vergani A., Mancia G.: Two novel nonsense mutations of HFE gene in five unrelated italian patients with hemochromatosis. Gastroenterology, 2000; 119: 441-445
[PubMed]
[91] Pointon J.J., Wallace D., Merryweather-Clarke A.T., Robson K.J.: Uncommon mutations and polymorphisms in the hemochromatosis gene. Genet. Test., 2000; 4: 151-161
[PubMed]
[92] Ponka P.: Iron metabolism: Physiology and pathophysiology. Trace. Elem. Res. Hum., 1999; 5: 55-57
[93] Roetto A., Daraio F., Porporato P., Caruso R., Cox T.M., Cazzola M., Gasparini P., Piperno A., Camaschella C.: Screening hepcidin for mutations in juvenile hemochromatosis: identification of a new mutation (C70R). Blood, 2004; 103: 2407-2409
[PubMed] [Full Text HTML] [Full Text PDF]
[94] Roetto A., Merryweather-Clarke A.T., Daraio F., Livesey K., Pointon J.J., Barbabietola G., Piga A., Mackie P.H., Robson K.J., Camaschella C.: A valine deletion of ferroportin 1: a common mutation in hemochromastosis type 4. Blood, 2002; 100: 733-734
[PubMed] [Full Text HTML] [Full Text PDF]
[95] Roetto A., Papanikolau G., Politou M., Alberti F., Gitrelli D., Christakis J., Loukopoulos D., Camaschella C.: Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet., 2003; 33: 21-22
[PubMed]
[96] Roetto A., Totaro A., Piperno A., Piga A., Longo F., Garozzo G., Cali A., De Gobbi M., Gasparini P., Camaschella C.: New mutations inactivating ransferrin receptor 2 in hemochromatosis type 3. Blood, 2001; 97: 2555-2560
[PubMed] [Full Text HTML] [Full Text PDF]
[97] Rosmorduc O., Poupon R., Nion I., Wendum D., Feder J., Bereziat G., Hermelin B.: Differential HFE allele expression in hemochromatosis heterozygotes. Gastroenterology, 2000; 119: 1075-1086
[PubMed]
[98] Roy C.N., Enns C.A.: Iron homeostasis: new tales from the crypt. Blood, 2000; 96: 4020-4027
[PubMed] [Full Text HTML] [Full Text PDF]
[99] Schimanski L.M., Drakesmith H., Merryweather-Clarke A.T., Viprakasit V., Edwards J.P., Sweetland E., Bastin J.M., Cowley D., Chinthammitr Y., Robson K.J., Townsend A.R.: In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood, 2005; 105: 4096-4102
[PubMed]
[100] Sham R.L., Phatak P.D., West C., Lee P., Andrews C., Beutler E.: Autosomal dominant hereditary hemochromatosis associated with a novel ferroportin mutation and unique clinical features. Blood Cells Mol. Dis., 2005; 34: 157-161
[PubMed]
[101] Sheldon J.H.: Haemochromatosis. Oxford University Press, 1935
[102] Simon M., Bourel M., Fauchet R., Genetet B.: Association of HLA-A3 and HLA-B14 antigens with idiopathic haemochromatosis. Gut, 1976; 17: 332-334
[PubMed]
[103] Touret N., Furuya W., Forbes J., Gros P., Grinstein S.: Dynamic traffic through the recycling compartment couples the metal transporter Nramp2 (DMT1) with the transferrin receptor. J. Biol. Chem., 2003; 278: 25548-25557
[PubMed] [Full Text HTML] [Full Text PDF]
[104] Trinder D., Olynyk J.K., Sly W.S., Morgan E.H.: Iron uptake from plasma transferrin by the duodenum is impaired in the HFE knockout mouse. Proc Natl Acad Sci U S A., 2002; 99: 5622-5626
[PubMed] [Full Text HTML] [Full Text PDF]
[105] Vulpe C.D., Kuo Y.M., Murphy T.L., Cowley L., Askwith C., Libina N., Gitschier J., Anderson G.J.: Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet., 1999; 21: 195-199
[PubMed] [Full Text HTML] [Full Text PDF]
[106] Waheed A., Grubb J.H., Zhou X.Y., Tomatsu S., Fleming R.E., Costaldi M.E., Britton R.S., Bacon B.R., Sly W.S.: Regulation of transferrinmediated iron uptake by HFE, the protein defective in hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA, 2002; 99: 3117-3122
[PubMed] [Full Text HTML] [Full Text PDF]
[107] Waheed A., Parkkila S., Zhou X.Y., Tomatsu S., Tsuchihashi Z., Feder J.N., Schatzman R.C., Britton R.S., Bacon B.R., Sly W.S.: Hereditary haemochromatosis: effects of the C282Y and H63D mutations on association with b
2-microglobulin, intracellular processing, and cell surface expression of the HFE protein in COS-7cells. Proc. Natl. Acad. Sci. USA, 1997; 94: 12384-12389
[PubMed] [Full Text HTML] [Full Text PDF]
[108] Wallace D.F., Pedersen P., Dixon J.L., Stephenson P., Searle J.W., Powell L.W., Subramaniam V.N.: Novel mutation in ferroportin1 is associated with autosomal dominant hemochromatosis. Blood, 2002; 100: 692-694
[PubMed] [Full Text HTML] [Full Text PDF]
[109] Wallace D.F., Summerville L., Lusby P.E., Subramaniam V.N.: First phenotypic description of transferrin receptor 2 knockout mouse, and the role of hepcidin. Gut, 2005; 54: 980-986
[PubMed]
[110] Wessling-Resnick M.: Biochemistry of iron uptake. Crit. Rev. Biochem. Mol. Biol., 1999; 34: 285-314
[PubMed]
[111] West A.P.Jr, Bennett M.J., Sellers V.M., Andrews N.C., Enns C.A., Bjorkman P.J.: Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J. Biol. Chem., 2000; 275: 38135-38138
[PubMed] [Full Text HTML] [Full Text PDF]
[112] Wigg A.J., Harley H., Casey G.: Heterozygous recipient and donor HFE mutations associated with a hereditary haemochromatosis phenotype after liver transplantation. Gut, 2003; 52: 433-435
[PubMed] [Full Text HTML] [Full Text PDF]
[113] Yip R.: Iron deficiency. Bull. World Health Organ., 1998; 76, Suppl.2: 121-123
[PubMed]
[114] Yoshida K., Furihata K., Takeda S., Nakamura A., Yamamoto K., Morita H., Hiyamuta S., Ikeda S., Shimizu N., Yanagisawa N.: A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat. Genet., 1995; 9: 267-272
[PubMed]
[115] Zaahl M.G., Merryweather-Clarke A.T., Kotze M.J., van der Merwe S., Warnich L., Robson K.J.: Analysis of genes implicated in iron regulation in individuals presenting with primary iron overload. Hum. Genet., 2004; 115: 409-417
[PubMed]
[116] Zhang A.S., Xiong S., Tsukamoto H., Enns C.A.: Localization of iron metabolism-related mRNAs in rat liver indicate that HFE is expressed predominantly in hepatocytes. Blood, 2004; 103: 1509-1514
[PubMed] [Full Text HTML] [Full Text PDF]