
Możliwości rozpoznawania i leczenia guzów neuroendokrynnych przewodu pokarmowego
Marek Bolanowski 1 , Beata Kos-Kudła 2Streszczenie
Guzy neuroendokrynne przewodu pokarmowego (GEP) są rzadkimi nowotworami, charakteryzującymi się zdolnością do syntezy, magazynowania i uwalniania hormonów i amin biogennych. Ze względu na rzadkie występowanie istnieją ograniczone dane kliniczne i prognostyczne o nich. Mogą dawać objawy kliniczne zależne od wydzielanych substancji, masy guza lub przerzutów. Oznaczanie swoistych i nieswoistych markerów guzów GEP charakteryzuje się wysoką czułością w ustaleniu właściwego rozpoznania, ma również istotne znaczenie w prognozowaniu przebiegu choroby. W diagnostyce obrazowej stosowane są endoskopowa ultrasonografia, tomografia komputerowa, rezonans magnetyczny, a zwłaszcza scyntygrafia z użyciem analogów somatostatyny. Radykalnym leczeniem guzów GEP jest postępowanie chirurgiczne, ale rzadko jest ono możliwe. Leczeniem z wyboru chorych z charakterystycznymi zespołami objawów klinicznych guzów GEP są analogi somatostatyny. W niskozróżnicowanych guzach lub w przypadkach zaawansowanej lub szybko postępującej choroby można stosować interferon alfa, chemioterapię i/lub leczenie radio-metaboliczne oparte na znakowanych radioznacznikami analogach somatostatyny. Przydatne jest także leczenie objawowe.
Słowa kluczowe: analogi somatostatyny • chemioterapia • guzy neuroendokrynne • guzy żołądkowo-jelitowo-trzustkowe • interferon alfa • rakowiak • rozpoznawanie • terapia radioizotopowa
Summary
Neuroendocrine tumors of the gastroenteropancreatic (GEP) system are rare neoplasms, characterized by their capacity to synthesize, store, and release hormonal products and biogenic amines. Because of their low incidence, limited data about clinical outcomes and prognostic variables are available. They can present clinical symptoms caused by the products secreted, tumor mass or metastases. Assessment of specific or nonspecific tumor markers offers high sensitivity in establishing diagnosis and can also have prognostic significance. Imaging modalities include endoscopic ultrasonography, computerized tomography, magnetic resonance imaging, and in particular, scintigraphy with somatostatin analogs. The radical treatment of GEP tumors is tumor surgery, but this is rarely possible. Somatostatin analogs have been the treatment of choice in symptomatic patients with GEP tumors. In poorly differentiated tumors or in selected cases of advanced or rapidly growing disease, interferon alpha, chemotherapy, and/or radio-metabolic treatment with radiolabeled somatostatin analogs can be performed. Symptomatic therapy is also helpful.
Key words: carcinoid • chemotherapy • diagnostics • gastroenteropancreatic tumors • interferon-alpha • neuroendocrine tumors • radionuclide therapy • somatostatin-analogs
Guzy neuroendokrynne przewodu pokarmowego, żołądkowo-jelitowo-trzustkowe (gastroenteropancreatic tumors – GEP) to guzy wywodzące się z gruczołów wewnętrznego wydzielania, grup komórek endokrynnych umiejscowionych wewnątrz tkanek gruczołowych oraz komórek endokrynnych rozproszonych pośród komórek egzokrynnych przewodu pokarmowego [16]. Mogą występować w całym organizmie. Opisywane dawniej pochodzenie guzów neuroendokrynnych (neuroendocrine tumors – NET) z komórek układu APUD (amine precursor uptake and decarboxylation) przypisywało tej różnorodnej grupie taki sam biologiczny fenotyp, co nie odzwierciedlało biologicznej i klinicznej heterogenności tych nowotworów [6].
Zapadalność na te nowotwory wynosi 13 przypadków na 1 000 000 mieszkańców na rok. Guzy typu GEP stanowią 70% wszystkich guzów NET oraz 2% wszystkich nowotworów przewodu pokarmowego. Występują z podobną częstością u obu płci, szczyt zapadalności przypada na 6. dekadę życia [29,38]. Najczęstszymi z nich są rakowiaki (około 50% wszystkich GEP). Rakowiaki, tradycyjnie klasyfikuje się na podstawie ich pochodzenia embrionalnego: z proksymalnego (foregut), środkowego (midgut) oraz dystalnego (hindgut) odcinka cewy jelitowej (foregut – układ oddechowy, grasica, żołądek, dwunastnica, trzustka; midgut – jelito cienkie, wyrostek robaczkowy, wstępnica, i hindgut – poprzecznica, esica, odbytnica) [23,26,38]. Zespół rakowiaka to grupa objawów klinicznych towarzyszących obecności rakowiaka i związanych z wydzielaniem serotoniny. Serotonina (5-HT) jest syntetyzowana z tryptofanu, przez prekursor 5-HTP, a jej metabolitem jest kwas 5-hydroksyindolooctowy (5-HIAA) – oznaczany w moczu. Gdy serotonina i inne produkty są wydzielane do krążenia wrotnego, podlegają metabolizmowi wątrobowemu i nie dochodzi do wystąpienia ogólnych objawów klinicznych. Natomiast, gdy dochodzi do przerzutów do wątroby, lub ognisko pierwotne znajduje się w oskrzelu lub jajniku, objawy zespołu rakowiaka są bardziej zaznaczone [16]. Do zespołu objawów rakowiaka zalicza się: zaczerwienienie skóry (flush), biegunkę, zmiany zastawkowe w prawej części serca, skurcze mięśni, teleangiektazje, obrzęki, sinicę, objawy bronchospastyczne, bóle brzucha, miopatię, sapanie, pigmentację skóry, objawy stawowe [36]. Przełom rakowiaka to zagrażające życiu powikłanie rakowiaka (najczęściej typu foregut i midgut) w postaci spadku ciśnienia tętniczego, tachyarytmii, świszczącego oddechu, zaczerwienienia skóry i zaburzeń czynności ośrodkowego układu nerwowego. Może wystąpić samoistnie lub częściej w następstwie znieczulenia ogólnego, interwencji chirurgicznej, leków (chemioterapia, radiofarmaceutyki) powodujących nagłe uwolnienie dużej ilości amin do krążenia ogólnego [16,38].
Rakowiaki są najczęściej umiejscowione w ścianie jelita, trzustce, wyrostku robaczkowym, poza tym w okrężnicy, odbytnicy, żołądku, dwunastnicy, płucu, oskrzelu, jajniku czy też grasicy. Często pierwszymi sygnałami rakowiaka są objawy związane z obecnością przerzutów. Guzy wielkości <1 cm dają przerzuty w 15%, podczas gdy rakowiaki większe niż 2 cm nawet w 95%. Objawy mogą występować epizodycznie, zwykle mija około 10 lat od pierwszych symptomów klinicznych do właściwego rozpoznania rakowiaka. Zespół objawów rakowiaka występuje u mniej niż 10% chorych z rakowiakiem, częściej w guzach jelita cienkiego [38]. Najnowsza klasyfikacja proponuje ograniczenie stosowania określenia rakowiaka wyłącznie do guzów wydzielających serotoninę, wywodzących się ze środkowego odcinka cewy jelitowej (midgut) [17].
Guzy GEP mogą być czynne bądź nieczynne wydzielniczo. Mogą wydzielać aminy biogenne, hormony eutopowe, lecz także ektopowo (np. rakowiak oskrzela wydzielający GHRH objawiający się akromegalią lub wydzielający kortykotropinę – ACTH, powodujący zespół Cushinga) [5,21]. Mogą występować sporadycznie lub w przebiegu zespołów mnogiej gruczolakowatości wewnątrzwydzielniczej (multiple endocrine neoplasia) MEN-I i MEN-II [36].
Objawy kliniczne zależą od substancji wydzielanych przez guzy [19,32,38]. Są to najczęściej objawy zespołu rakowiaka zależne od wydzielania serotoniny, innymi wydzielanymi substancjami mogą być ACTH, histamina, dopamina, neurotensyna, substancja P, kalikreina, prostaglandyny i tachykininy [20,21]. Guzy GEP rzadko (<1%) się objawiają nieprawidłową glikemią (hipo-, lub hiperglikemią), chorobą wrzodową, osutką skóry. Są one w większości złośliwe (z wyjątkiem insulinoma), jednak stopień ich złośliwości jest bardzo zróżnicowany w zależności od rodzaju guza. Mogą dawać przerzuty drogą krwi i chłonki do wątroby, kości, węzłów chłonnych. Przerzuty do kości i wątroby są najczęstszą przyczyną zgonu pacjentów, przeżycie 5-letnie osiąga mniej niż 50% tych chorych. Niekorzystne rokowniczo są ponadto wiek powyżej 50 r.ż, płeć męska, umiejscowienie zmiany w trzustce bądź jelicie grubym, wielkość zmiany i głębokość wniknięcia tkanek, obecność objawów klinicznych w chwili rozpoznania. Podobnie, rokowanie pogarszają niemożność leczenia operacyjnego, duże stężenie markerów aktywności choroby (chromograniny, 5-HIAA, gastryna, ACTH) oraz wysokie wartości wskaźników proliferacyjnych [16,36].
KLINICZNA CHARAKTERYSTYKA GUZÓW GEP
Insulinoma – najczęstszy wyspiak trzustki, wywodzi się z komórek beta wysp trzustkowych, wydziela insulinę, powoduje hipoglikemię, osłabienie, drżenia, utraty świadomości. Ten wyspiak rzadko cechuje się złośliwym przebiegiem (10%), w takim samym odsetku może występować pod postacią mnogich ognisk [12,23,34].
Gastrinoma – guz trzustki lub dwunastnicy wydzielający gastrynę, powoduje nadkwasotę, objawia się nawracającą chorobą wrzodową żołądka i biegunką (zespół Zollingera- Ellisona). W 5% przypadków występuje w nietypowych miejscach. Gastrinoma może wydzielać ACTH prowadząc do zespołu Cushinga. W 1/3 przypadków gastrinoma wchodzi w skład zespołu MEN-I. Sporadycznie występujące gastrinoma bywają w 40–80% złośliwe [12,32,38].
VIP-oma – wywodzi się z komórek autonomicznego układu nerwowego, może być umiejscowiony w trzustce, układzie nerwowym lub nadnerczach. Wydzielanie wazoaktywnego polipeptydu jelitowego VIP (vasoactive intestinal polypeptide) powoduje uporczywą wodnistą biegunkę, hipokalemię, achlorhydrię – zespół Vernera-Morrisona [32,38].
PP-oma – guz wydzielający polipeptyd trzustkowy (pancreatic polypeptide – PP), tego typu guzy są przeważnie bezobjawowe, mogą się objawiać biegunką, utratą masy ciała, rzadko cukrzycą [37].
Glukagonoma – wyspiak wywodzący się z komórek alfa i prowadzący do nekrolitycznego rumienia skóry o charakterze pełzającym okolicy ust i genitalii, łagodnej cukrzycy, utraty masy ciała i częstych epizodów zatorowo- zakrzepowych. Bywa przyczyną depresji, zwłaszcza u kobiet [35,38].
Somatostatinoma – wyspiak z komórek D, wydziela somatostatynę (SS), występuje częściej u kobiet. Jego objawami są stolce tłuszczowe, kamica pęcherzyka żółciowego, bóle brzucha, cukrzyca, hipochlorhydria, zaburzenia czynnościowe pęcherzyka żółciowego, utrata masy ciała. Guzy wywodzące się z trzustki są duże (około 5 cm) – dominują wtedy objawy miejscowe guza [16,37].
Neurotensinoma – guz wydzielający neurotensynę, objawia się nadciśnieniem, tachykardią, sinicą, obrzękami, rozszerzeniem naczyń, cukrzycą [37].
Grelinoma (ghrelinoma) – wydzielanie greliny może się wiązać z hiperglikemią, niedoborem insuliny, insulinoopornością, zaburzeniami motoryki jelit [7,38].
Rzadko mogą być wydzielane inne substancje, np.: ACTH, GHRH, parathormon, kalcytonina, enteroglukagon, cholecystokinina, neurokininy itp. [16,32,38]. Guzy niewydzielające (nieczynne hormonalnie) – mogą dawać niecharakterystyczne objawy: bóle brzucha, żółtaczkę mechaniczną, zaburzenia motoryki przewodu pokarmowego [16].
Receptory somatostatynowe
Znaczna część guzów NET ma dużą liczbę receptorów SS (80% wykazuje ekspresję receptora typu 2) i dlatego możliwe jest uwidocznienie ich za pomocą analogu somatostatyny – oktreotydu znakowanego indem 111 [4,25].
KLASYFIKACJA HISTOPATOLOGICZNA
Większość czynnych guzów NET wytwarza, magazynuje oraz wydziela peptydy i aminy biogenne. Badanie histopatologiczne pozwala na klasyfikację tych guzów według ich pochodzenia tkankowego, charakteru biochemicznego i przewidywanego rokowania choroby. Metody badania obejmują zarówno diagnostykę za pomocą mikroskopu świetlnego i elektronowego, jak i badania histochemiczne i immunohistochemiczne. Najczęściej stosowanymi markerami cytosolowymi są: neuronoswoista enolaza (neuronspecific enolase – NSE) i PGP 9.5 (protein gene product 9.5), a markerami ziarninowymi: chromogranina A (CgA) i synaptofizyna [16].
Światowa Organizacja Zdrowia (WHO) zaproponowała w 2000 r. następującą klasyfikację histopatologiczną guzów NET:
1. Dobrze zróżnicowane guzy endokrynne (łagodne lub o niskim stopniu złośliwości).
2. Dobrze zróżnicowane raki endokrynne.
3. Słabo zróżnicowane raki endokrynne (rak drobnokomórkowy).
4. Mieszane raki egzokrynno-endokrynne [17].
Różnicowanie opiera się na cechach histomorfologicznych guza, jego rozmiarach i obecności miejscowego naciekania lub/i przerzutów, które odzwierciedlają charakter biologiczny nowotworu [16]. Ponadto powinno się również zwrócić uwagę na wiele innych parametrów, takich jak: naciekanie naczyń, obecność martwicy, zwiększenie wskaźnika proliferacyjnego Ki-67 powyżej 2%, które są istotne dla późniejszego rokowania [6,27]. Stworzona w ostatnim czasie nowa klasyfikacja ograniczyła stosowanie określenia rakowiak, najczęstszego z guzów GEP, tylko do guzów wydzielających serotoninę, wywodzących się ze środkowego odcinka cewy jelitowej. Nadal jednak chętnie opieramy się na tradycyjnym podziale rakowiaków (foregut, midgut i hindgut) [26]. Podział ten, z zaznaczeniem charakterystycznych cech w badaniu histopatologicznym, w diagnostyce molekularnej wraz z najczęstszymi produktami wydzielania rakowiaków, przedstawia tabela 1 [19,26].
Tabela 1. Tradycyjna klasyfikacja rakowiaków
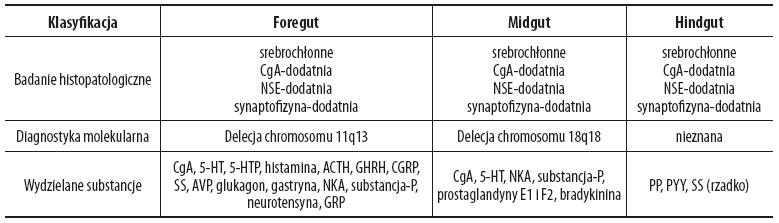
ACTH – adrenokortykotropina; AVP – arginino-wazopresyna; CgA – chromogranina A; CGRP – peptyd zależny od genu kalcytoniny; GHRH – hormon uwalniający hormon wzrostu; GRP – peptyd uwalniający gastrynę; 5-HT – 5 hydroksytryptamina; 5-HTP – 5 hydroksytryptofan; NKA – neurokinina; NSE – neuronoswoista enolaza; PP – polipeptyd trzustkowy; PYY – peptyd YY, SS – somatostatyna. Zmodyfikowano wg [26].
Swoiste markery guzów neuroendokrynnych
Różne typy komórek czynnych hormonalnie guzów wydzielają swoiste peptydy, aminy biogenne i hormony, które mogą być wskaźnikami użytecznymi w diagnostyce i monitorowaniu leczenia tych nowotworów. Bezpośredni pomiar ich stężeń pozwala ustalić diagnozę i pośrednio szacować wielkość guza. Niektóre guzy mogą wydzielać jednocześnie kilka hormonów, a ich stężenia mogą się zmieniać w przebiegu choroby i pod wpływem terapii [9].
W rozpoznawaniu zespołu rakowiaka najbardziej użytecznym badaniem jest wydalanie 5-HIAA w moczu. Fałszywie dodatnie wyniki oznaczeń można zaobserwować u chorych z zespołami złego wchłaniania. Badanie może natomiast dać wyniki ujemne u pacjentów z rakowiakiem oskrzeli i żołądka (foregut), u których często brak jest dekarboksylazy aminokwasów aromatycznych. W wątpliwych przypadkach przydatne może być oznaczenie stężenia serotoniny we krwi [26].
Biochemiczne markery guzów neuroendokrynnych
W diagnostyce guzów NET wykorzystywane są również nieswoiste markery, takie jak chromogranina A (CgA), NSE oraz podjednostki a
i b
ludzkiej gonadotropiny kosmówkowej (hCG). Te ostatnie są też wykorzystywane w diagnostyce guzów GEP hormonalnie nieczynnych [16]. Chromograniny (oznaczane jako A, B i C) są białkami wytwarzanymi, magazynowanymi i uwalnianymi z tkanek neuroendokrynnych. Najwyższe stężenia CgA wykazano u chorych z rakowiakiem z przerzutami i innymi guzami GEP. W interpretacji wyników CgA należy jednak wziąć pod uwagę to, że czułość i swoistość oznaczeń waha się odpowiednio pomiędzy 10–100% oraz 68–100%. CgA jest natomiast ważnym markerem w monitorowaniu przebiegu choroby i leczenia guzów GEP oraz niezależnym czynnikiem prognostycznym przeżycia u chorych z rakowiakiem typu midgut. Innym istotnym wskaźnikiem oceny stopnia złośliwości guzów GEP jest antygen CA-19 [16,24,36]. W praktyce klinicznej jest zalecana ocena stężenia CgA w odniesieniu do innych wskaźników charakterystycznych dla danego zespołu, np. 5-HIAA (w zespole rakowiaka) czy gastryny (w gastrinoma). Guzy nieczynne hormonalnie mogą być diagnozowane z wykorzystaniem oznaczeń CgA oraz dodatkowo takich wskaźników, jak PP czy a
-hCG [27].
Jeśli u chorych z charakterystycznymi objawami klinicznymi stwierdza się prawidłowe wartości swoistych markerów dla danego zespołu a istnieje duże podejrzenie występowania guza GEP, można wtedy wykonać testy stymulacyjne, np. test pobudzenia sekretyną lub dożylnym wlewem wapnia w przypadku gastrinoma. W najnowszym piśmiennictwie przydatność kliniczna tych testów jest kwestionowana [16,36].
BADANIA OBRAZOWE SŁUŻĄCE UMIEJSCOWIENIU GUZÓW GEP
Badanie ultrasonograficzne (USG)
Szczególną rolę w diagnostyce lokalizacyjnej guzów GEP przypisuje się endoskopowej ultrasonografii (EUS). Ostatnie badania wykazały 93% czułość i 95% swoistość tej metody w obrazowaniu guzów GEP zlokalizowanych w trzustce [3]. Wykorzystując metodę wewnątrzprzewodowej EUS lokalizuje się zmiany wewnątrz przewodu trzustkowego w trakcie endoskopowej choledochopankreatografii wstecznej (ERCP) za pomocą ultracienkiej sondy o średnicy 2 mm. Z kolei, śródoperacyjne techniki lokalizacyjne, polegające na badaniu USG połączonym z palpacją narządu dają czułość 58–100% [13].
Tomografia komputerowa (TK) i rezonans magnetyczny (MR)
Czułość spiralnej TK z użyciem środka kontrastowego dla guzów trzustki wynosi 82–92%. Guzy GEP nieczynne hormonalnie oraz VIP-oma i glukagonoma zazwyczaj osiągają wymiary ponad 3 cm, dlatego w chwili rozpoznania czułość TK sięga 100%. TK jest też badaniem z wyboru w diagnostyce przerzutów do wątroby i kości u chorych z guzami NET [16]. Badanie MR jest uznawane za porównywalne z TK w dokładności rozpoznawania guzów GEP, może być wykorzystywane w monitorowaniu odpowiedzi na leczenie [8]. Nowością jest zastosowanie swoistego dla wątroby kontrastu (trójsodowy mangafodipir – Mn-DPDP), umożliwiającego uwidocznienie przerzutów guzów NET [40].
Scyntygrafia receptorów somatostatyny
Guzy mające receptory somatostatyny (SS) mogą być uwidocznione w scyntygrafii za pomocą oktreotydu znakowanego 111indem (OctreoScan) [25]. Metoda ta jest szczególnie skuteczna w obrazowaniu guzów typu gastrinoma, glukagonoma, nieczynnych guzów trzustki i rakowiaka [22]. U chorych z rakowiakiem scyntygrafia ze znakowanym indem pentetreotydem jest czulsza niż starsza technika oparta na stosowaniu 123meta-jodobenzyloguanidyny (MIBG) i pozwala na zidentyfikowanie większości guzów nowotworowych [14]. Połączenie techniki tomografii komputerowej emisji pojedynczego fotonu (SPECT) 111Inoktreotydu z warstwowymi obrazami TK/MR pozwala na polepszenie możliwości lokalizacji i oceny guzów neuroendokrynnych (obraz anatomiczno-czynnościowy) [15]. Rozmieszczenie receptorów SS u chorych z guzami NET przedstawia tabela 2 [16,19].
Tabela 2. Markery biochemiczne i rozmieszczenie receptorów dla somatostatyny (SS) u chorych z guzami NET
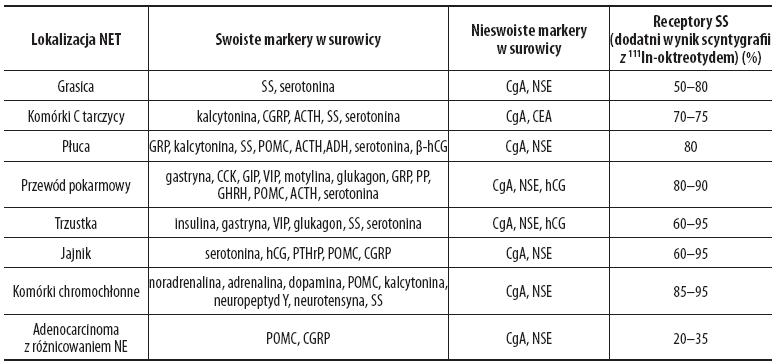
ACTH – adrenokortykotropina; ADH – wazopresyna; hCG – ludzka gonadotropina kosmówkowa; CgA – chromogranina A; CCK – cholecystokinina; CGRP – peptyd zależny od genu kalcytoniny; GIP – żołądkowy polipeptyd hamujący; GRP – peptyd uwalniający gastrynę; NSE – neuronoswoista enolaza; POMC – proopiomelanokortyna; PP – polipeptyd trzustkowy; PTHrP – peptyd zależny od parathormonu; SS – somatostatyna; VIP – wazoaktywny peptyd jelitowy. Zmodyfikowano wg [16].
Pozytronowa tomografia emisyjna (PET)
Badanie PET wykorzystuje izotopy o krótkim okresie połowicznego rozpadu, np. analogi somatostatyny znakowane miedzią (64Cu-TETA-oktreotyd). Charakteryzuje się ona wysoką czułością i swoistością, co czyni ją obiecującą techniką w lokalizacji guzów neuroendokrynnych [2].
Angiografia z pobieraniem próbek krwi żylnej po stymulacji tętniczej
Ta inwazyjna metoda zarezerwowana jest do trudnych przypadków, szczególnie guzów trzustki, gdy nie udaje się zlokalizować guza innymi metodami. Czułość angiografii zwiększa podawanie do tętnic trzustki substancji stymulujących (np. sekretyny w przypadku gastrinoma i glukonianu wapnia w przypadku insulinoma) z pobieraniem próbek krwi żylnej z wątroby [12,16].
MOŻLIWOŚCI TERAPEUTYCZNE
Leczenie operacyjne
Leczeniem z wyboru guzów NET jest leczenie operacyjne. W większości przypadków całkowite usunięcie chirurgiczne zmiany nowotworowej jest jednak niemożliwe ze względu na zaawansowanie procesu chorobowego. Ostatnio, jako postępowanie wspomagające, są często wykorzystywane operacje cytoredukcyjne. Polegają one na resekcji guza, ablacyjnej radioterapii i krioterapii, mających na celu zatrzymanie wzrostu nowotworu, poprawę objawów klinicznych i utrzymanie dobrej jakości życia chorych [27]. Wybiórcza embolizacja i chemoembolizacja tętnic wątrobowych mogą stanowić skuteczne uzupełnienie leczenia operacyjnego [1,30].
Farmakoterapia
Analogi somatostatyny
Przełomem w leczeniu objawowym hormonalnie czynnych guzów GEP stało się zastosowanie analogów SS, szczególnie postaci o przedłużonym działaniu. SS ma wielokierunkowy wpływ na przewód pokarmowy przez hamowanie wydzielania hormonów trzustkowych i jelitowych, m.in. insuliny, glukagonu, gastryny, sekretny i VIP, a także hamowanie jego motoryki i transportu jelitowego, przepływu krwi w naczyniach trzewnych oraz wzrostu i różnicowania tkanek. Natywna SS ma krótki okres półtrwania, podobnymi właściwościami charakteryzują się krótkodziałające analogi SS (oktreotyd).
SS i jej analogii wywierają swoje biologiczne działanie poprzez wiązanie się z receptorami SS (podtypy SS 1-5), przy czym oktreotyd wykazuje największe powinowactwo do receptora podtypu 2, mniejsze zaś do podtypów 3 i 5. Analogi SS (oktreotyd i lanreotyd) są wykorzystywane do opanowania objawów klinicznych zależnych od nadmiernej sekrecji hormonów wydzielanych przez guzy GEP z ekspresją receptorów SS 2 i SS 5 [8,33,42]. Przeciwnowotworowe działanie analogów SS może odbywać się w sposób bez- pośredni poprzez wpływ na receptory somatostatynowe obecne na błonach komórkowych guza, jak również pośrednio przez hamowanie wydzielania wielu czynników wzrostu, np. insulinopodobnego czy naskórkowego czynnika wzrostu (IGF-1, EGF), modulujący wpływ na układ immunologiczny oraz indukcję apoptozy i hamowanie angiogenezy [28].
Zastosowanie analogów SS poprawiło znacznie rokowanie w zespole rakowiaka. Preparaty oktreotydu, zarówno krótko-, jak i długodziałające, skutecznie kontrolują objawy napadowych zaczerwienień skóry, tzw. flush, biegunek oraz zmniejszają wydalanie 5-HIAA u chorych z zespołem rakowiaka. Poprawę objawów klinicznych po zastosowaniu oktreotydu LAR lub lanreotydu obserwowano u 30–85% chorych, a obniżenie stężeń markerów guza w około 50% przypadków. Wykazano jednak niewielki wpływ takiego leczenia na zmniejszenie masy guza. Badania z zastosowaniem analogów o przedłużonym działaniu potwierdziły regresję guza jedynie w 5% przypadków, natomiast stabilizację wzrostu guza u 40–80% chorych. Analogi SS są też lekami z wyboru w leczeniu przełomu rakowiaka [11,26,28,41].
Przedmiotem badań, zarówno eksperymentalnych, jak i klinicznych są nowe analogi SS. Duże nadzieje wiąże się z analogami SOM 230 i KE 108, które charakteryzują się unikalnym profilem wiązania, łączą się bowiem ze wszystkimi podtypami receptorów SS 1–5 z podobnym powinowactwem i wydają się obiecującym narzędziem walki w terapii guzów NET wykazujących ekspresję wielu podtypów receptora SS [28]. Inną obiecującą terapią z wykorzystaniem analogów SS są skoniugowane analogi o działaniu cytotoksycznym, których przedstawicielem jest AN-238, będący połączeniem analogu SS z pirolinodoksorubicyną. Ten rodzaj celowanego leczenia, będący skutecznym modelem selektywnej chemioterapii, wykorzystuje swoiste wiązanie analogu SS z receptorem SS 2 na powierzchni komórki guza, jednocześnie dostarczając bezpośrednio do komórki czynnik cytotoksyczny i w ten sposób redukując obwodową toksyczność chemioterapeutyku. To bezpośrednie działanie skierowane przeciwko komórkom guza okazało się efektywne w modelach doświadczalnych guzów okrężnicy, jajnika, trzustki i prostaty. Brak jest jednak na razie odpowiednich badań klinicznych u chorych z guzami GEP [28].
Interferon a
Przeciwnowotworowe działanie interferonu alfa (INF-a
) polega na bezpośrednim wpływie na proliferację, apoptozę, różnicowanie i angiogenezę. INF-a
działa również immunomodulująco oraz indukuje procesy włóknienia w przerzutach, głównie do wątroby. Wyniki przeprowadzonych badań u chorych z rakowiakiem i guzami GEP umiejscowionymi w trzustce dowodzą, że przeciętna odpowiedź biochemiczna i opanowanie objawów klinicznych wynosi odpowiednio 40–70% oraz 44%, przy czym wpływ na zmniejszenie masy guza występuje tylko w 11% [16].
Lepszych wyników można oczekiwać w następstwie łącznej terapii analogami SS i INF-a
. Należy się spodziewać zmniejszenia nasilenia (ustąpienia) objawów klinicznych, poprawy odpowiedzi biochemicznej, a także zahamowania wzrostu guza i prawdopodobnie przedłużenia czasu przeżycia w grupach chorych odpowiadających na leczenie [16]. W randomizowanych prospektywnych badaniach u chorych z rozsianym rakowiakiem, po przeprowadzonym pierwotnym leczeniu chirurgicznym z embolizacją tętnicy wątrobowej, wykazano, że dołączenie INF-a
do oktreotydu powoduje istotną redukcję progresji guza, jednak bez wpływu na czas przeżycia chorych [10]. Z kolei w innym wieloośrodkowym, randomizowanym, prospektywnym badaniu udowodniono, że stosowanie lanreotydu, INF-a
, a także połączenia tych dwóch leków nie działało antyproliferacyjnie u chorych z guzami GEP z przerzutami, zarówno czynnymi, jak i nieczynnymi hormonalnie. Odpowiedź na leczenie była mniejsza niż we wcześniej publikowanych, nierandomizowanych badaniach [18]. Problem ten wymaga więc dalszych wyjaśnień.
Chemioterapia
Większość guzów NET słabo reaguje na chemioterapię. Wynika to z biologicznego charakteru tych nowotworów, a szczególnie z ich niskiego indeksu mitotycznego. Badania nowych leków stosowanych w systemowej chemioterapii są zaś utrudnione ze względu na rzadkie występowanie NET oraz ich biologiczną i kliniczną różnorodność. Leczenie cytotoksyczne (m.in. 5-fluorouracyl, doksorubicyna, cisplatyna) jest standardem u chorych z guzami endokrynnymi trzustki, natomiast jest mniej skuteczne u chorych z rakowiakiem [28].
Różne rodzaje chemioterapii były dotychczas podawane w dobrze zróżnicowanych guzach GEP z niewielką skutecznością, szczególnie w rakowiakach. Chemioterapia nie może być więc leczeniem pierwszego rzutu u chorych z guzami GEP, powinna być zarezerwowana dla przypadków z zaawansowaną postacią choroby, przede wszystkim dla niskozróżnicowanych guzów trzustki [28].
Terapia radioizotopowa
Technika radioizotopowa jest wykorzystywana od wielu lat, głównie w leczeniu paliatywnym guzów GEP. Początkowo stosowano radioizotop 131I-MIBG i 125I-MIBG uzyskując około 30% odpowiedź na taką terapię u chorych z rakowiakiem. W poszukiwaniu zwiększenia skuteczności tego leczenia zauważono, że premedykacja nieznakowanym radioizotopowo MIBG powoduje zwiększony wychwyt 131I-MIBG, przedłuża działanie paliatywne oraz charakteryzuje się lepszą odpowiedzią biochemiczną [26,31]. W ostatnich kilku latach wprowadzono do leczenia również znakowane radioizotopami analogi SS: 111In-DTPA-oktreotyd i 90Y-DOTA-TOC. Efekt terapeutyczny znakowanego indem analogu 111In-DTPA-oktreotydu obejmuje częściową remisję, a przede wszystkim stabilizację pierwotnie zaawansowanego procesu nowotworowego [22]. Oparty na oktreotydzie radionuklid 90Y-DOTA-TOC, emitujący promieniowanie b
, wiąże się z receptorami typu 2 i 5 dla SS dając wysoki odsetek obiektywnych odpowiedzi na leczenie, wydłużenie czasu przeżycia i złagodzenie dolegliwości chorych z guzami NET [39]. Celowane leczenie znakowanymi radioizotopowo analogami SS wydaje się nieść istotne korzyści kliniczne. Trudnym jednak problemem wśród niepożądanych skutków tej terapii jest uszkodzenie nerek. Aby zapobiec nerkowej absorpcji peptydów stosuje się wlewy aminokwasów. Ta obiecująca postać celowanej terapii znakowanymi radioizotopowo analogami SS musi być jednak dokładnie oceniona w przyszłych badaniach klinicznych [15,25]. Zintegrowane z chemioterapią leczenie 131I-MIBG ma za zadanie zapobiec oporności klonów komórek niewychwytujących radioizotopu [15].
Inne możliwości farmakoterapii
W leczeniu objawowym insulinoma przydatne są diazoksyd i streptozotocyna – zmniejszające wydzielanie insuliny i skutecznie zapobiegające hipoglikemii. Mniejsze znaczenie mogą mieć werapamil, fenytoina, hormon wzrostu i glikokortykosteroidy. Wydzielanie kwasu solnego w zespole Zollingera-Ellisona mogą skutecznie zmniejszyć inhibitory pompy protonowej i leki blokujące receptory histaminowe H2, a leki przeciwbiegunkowe, indometacyna i lit mogą być przydatne w biegunkach w przebiegu VIPoma [16,38].
ROKOWANIE
Czynnikami niekorzystnymi rokowniczo w guzach GEP są wiek ponad 50 lat, płeć męska, lokalizacja w trzustce i odbytnicy, wielkość guza i głębokość penetracji tkankowej, obecność przerzutów i brak możliwości radykalnego leczenia. Rokowanie pogarszają także obecność objawów klinicznych, szczególnie objawów zespołu rakowiaka, wysokie wartości markerów (CgA, 5-HIAA, kalcytonina, gastryna, ACTH) oraz wysokie indeksy proliferacyjne [16,24,38].
Guzy neuroendokrynne wywodzące się z przewodu pokarmowego, to rzadko stwierdzane, małe i powoli rosnące guzy, bardzo trudne do zdiagnozowania. Ich właściwe rozpoznanie umożliwiające leczenie wymaga dużego stopnia „podejrzliwości” ze strony lekarza [36].
PIŚMIENNICTWO
[1] Ahlman H., Wangberg B., Jansson S., Friman S., Olausson M., Tylen U., Nilsson O.: Interventional treatment of gastrointestinal neuroendocrine tumours. Digestion, 2000; 62 (Suppl. 1): 59-68
[PubMed]
[2] Anderson C.J., Dehdashti F., Cutler P.D., Schwartz S.W., Laforest R., Bass L.A., Lewis J.S., McCarthy D.W.: 64Cu-TETA-octreotide as a PET imaging agent for patients with neuroendocrine tumors. J. Nucl. Med., 2001; 42: 213-221
[PubMed]
[3] Anderson M.A., Carpenter S., Thompson N.W., Nostrant T.T., Elta G.H., Scheiman J.M.: Endoscopic ultrasound is highly accurate and directs management in patients with neuroendocrine tumors of the pancreas. Am. J. Gastroenterol., 2000; 95: 2271-2277
[PubMed]
[4] Bertherat J., Tenenbaum F., Perlemoine K., Videau C., Alberini J.L., Richard B., Dousset B., Bertagna X., Epelbaum J.: Somatostatin receptors 2 and 5 are the major somatostatin receptors in insulinomas: an in vivo and in vitro study. J. Clin. Endocrinol. Metab., 2003; 88: 5353-5360
[PubMed] [Full Text HTML] [Full Text PDF]
[5] Bolanowski M., Schopohl J., Marciniak M., Rzeszutko M., Zatońska K., Daroszewski J., Milewicz A., Malczewska J., Badowski R.: Acromegaly due to GHRH-secreting large bronchial carcinoid. Complete recovery following tumor surgery. Exp. Clin. Endocrinol. Diabetes, 2002; 110: 188-192
[PubMed]
[6] Caplin M., Wiedenmann B.: The management of patients with neuroendocrine tumors. Endocr. Relat. Cancer, 2003; 10: 425-426
[7] Corbetta C., Peracchi M., Cappiello V., Lania A., Lauri E., Vago L., Beck-Peccoz P., Spada A.: Circulating ghrelin levels in patients with pancreatic and gastrointestinal neuroendocrine tumors: identification of one pancreatic ghrelinoma. J. Clin. Endocrinol. Metab., 2003; 88: 3117-3120
[PubMed] [Full Text HTML] [Full Text PDF]
[8] Debray M.P., Geoffroy O., Laissy J.P., Lebtahi R., Silbermann-Hoffman O., Henry-Fuegeas M.C., Cadiot G., Mignon M., Schouman-Claeys E.: Imaging, appearances of metastases from neuroendocrine tumours of the pancreas. Br. J. Radiol., 2001; 74: 1065-1070
[PubMed] [Full Text HTML] [Full Text PDF]
[9] Eriksson B., Šberg K., Stridsberg M.: Tumor markers in neuroendocrine tumors. Digestion, 2000; 62(Suppl. 1): 33-38
[PubMed]
[10] Faiss S., Pape U.F., Bohmig M., Dorffel Y., Mansmann U., Golder W., Riecken E.O., Wiedenmann B.: Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors – The International Lanreotide and Interferon Alfa Study Group. J. Clin. Oncol., 2003; 21: 2689-2696
[PubMed] [Full Text HTML] [Full Text PDF]
[11] Garland J., Buscombe J.R., Bouvier C., Bouloux P., Chapman M.H., Chow A.C., Reynolds N., Caplin M.E.: Sandostatin LAR (long-acting octreotide acetate) for malignant carcinoid syndrome: a 3-year experience. Aliment. Pharmacol. Ther., 2003; 17: 437-444
[PubMed] [Full Text HTML] [PubMed]
[12] Hammond P.J., Jackson J.A., Bloom S.R.: Localization of pancreatic tumours. Clin. Endocrinol., 1994; 40: 3-14
[13] Huai J.C., Zhang W., Niu H.O., Su Z.X., McNamara J.J., Machi J.: Localization and surgical treatment of pancreatic insulinomas guided by intraoperative ultrasound. Am. J. Surg., 1998; 175: 18-21
[PubMed]
[14] Kaltsas G., Korbonits M., Heintz E., Mukherjee J.J., Jenkins P.J., Chew S.L., Reznek R., Monson J.P., Besser G.M., Foley R., Britton K.E., Grossman A.B.: Comparison of somatostatin analog and meta-iodo-benzylguanidine radionuclides in the diagnosis and localization of advanced neuroendocrine tumors. J. Clin. Endocrinol. Metab., 2001; 86: 895-902
[PubMed] [Full Text HTML] [Full Text PDF]
[15] Kaltsas G., Rockall A., Papadogias D., Reznek R., Grossman A.B.: Recent advances in radiological and radionuclide imaging and therapy of neuroendocrine tumours. Eur. J. Endocrinol., 2004; 151: 15-27
[PubMed] [Full Text PDF]
[16] Kaltsas G.A., Besser G.M., Grossman A.B.: The diagnosis and medical management of advanced neuroendocrine tumors. Endocr. Rev., 2004; 25: 458-511
[PubMed]
[17] Kloppel G., Perren A., Heitz P.U.: The gastroenteropancreatic neuroendocrine cell system and its tumors: The WHO classification. Ann. NY Acad. Sci., 2004; 1014: 13-27
[PubMed]
[18] Kolby L., Persson G., Franzen S., Ahren B.: Randomized clinical trial of the effect of interferon alpha on survival in patients with disseminated midgut carcinoid tumours. Br. J. Surg., 2003; 90: 687-693
[PubMed]
[19] Kos-Kudła B.: Guzy neuroendokrynne. Endokrynologia Polska, 2004; 55: 492-499
[Full Text PDF]
[20] Kulke M.H., Mayer R.J.: Carcinoid tumors. N. Engl. J. Med., 1999; 340: 858-868
[Abstract]
[21] Limper A.H., Carpenter P.C., Scheithauer B., Staats B.A.: The Cushing syndrome induced by bronchial carcinoid tumors. Ann. Intern. Med., 1992; 117: 209-214
[PubMed]
[22] Maini C.L., Sciuto R., Festa A., Rea S., Romano L.: The role of nuclear medicine in GEP-NET diagnosis and therapy. W: Update in Neuroendocrinology (red. R. Baldelli, F.F. Casanueva, G. Tamburrano). Udine Centro UD, 2004; 529-544
[23] Monson J.P.: The epidemiology of endocrine tumours. Endocr. Relat. Cancer, 2000; 7: 29-36
[Full Text PDF]
[24] Nehar D., Lombard-Bohas C., Olivieri S., Claustrat B., Chayvialle J.-A., Penes M.-C., Sassolas G., Borson-Chazot F.: Interest of chromogranin A for diagnosis and follow-up of endocrine tumours. Clin. Endcocrinol., 2004; 60: 644-652
[PubMed] [Full Text HTML] [Full Text PDF]
[25] Oberg K.: Carcinoid tumors: molecular genetics, tumor biology, and update of diagnosis and treatment. Curr. Opin. Oncol., 2002; 14: 38-45
[PubMed]
[26] Oberg K.: Carcinoid tumors, carcinoid syndrome and related disorders. W: Williams Textbook of Endocrinology (red. P.R. Larsen). Saunders, Philadelphia, 2003; 1857-1874
[27] Oberg K., Kvols L., Caplin M., Delle Fave G., de Herder W., Rindi G., Ruszniewski P., Woltering E.A., Wiedenmann B.: Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann. Oncol., 2004; 15: 966-973
[PubMed]
[28] Panzuto F., Nansoni S., Corleto V.D., Delle Fave G.: Pharmacological treatment of gastroenteropancreatic neuroendocrine tumors. W: Update in Neuroendocrinology (red. R. Baldelli, F.F. Casanueva, G. Tamburrano). Udine Centro UD, 2004; 547-561
[29] Perri P., Cavaliere F., Botti C., Diacono F., Di Filippo F., Guadagni F., Bellantone R., Tamburrano G., Baldelli R.: Epidemiology of gastroenteropancreatic neuroendocrine tumors. W: Update in Neuroendocrinology (red. R. Baldelli, F.F. Casanueva, G. Tamburrano). Udine Centro UD, 2004; 483-512
[30] Ruszniewski P., Malka D.: Hepatic arterial chemoembolization in the management of advanced digestive endocrine tumors. Digestion, 2000; 62(Suppl. 1): 79-83
[PubMed]
[31] Taal B.G., Hoefnagel C., Boot H., Valdes O.R., Rutgers M.: Improved effect of 131I-MIBG treatment by predosing with non-radiolabeled MIBG in carcinoid patients, and studies in xenografted mice. Ann. Oncol., 2000; 11: 1437-1443
[PubMed]
[32] Taheri S., Ghatei M.A., Bloom S.R.: Gastrointestinal hormones and tumor syndromes. W: Endocrinology. Red. DeGroot L.J., Jameson J.L.), t. 3, W.B. Saunders Co., 2001; 2547-2558
[33] Tomassetti P., Migliori M., Gullo L.: Slow-release lanreotide treatment in endocrine gastrointestinal tumors. Am. J. Gastroenterol., 1998; 93: 1468-1471
[PubMed]
[34] Tso A.W.K., Lam K.S.M.: Insulinoma. Curr. Opin. Endocrinol. Diabetes, 2000; 7: 83-86
[35] van Beek A.P., de Haas E.R.M., van Vloten W.A., Lips C.J.M., Roijers J.F.M., Canninga-van Dijk M.R.: The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur. J. Endocrinol., 2004; 151: 531-537
[PubMed] [Full Text PDF]
[36] Vinik A.: Neuroendocrine tumors. The Endocrine Society’s 86 Annual Meeting: ENDO 2004, 16-19 June, New Orleans, Meet-the-Professor Handouts, 233-246
[37] Vinik A.I., Strodel W.E., Eckhauser F.E., Moattari A.R., Lloyd R.: Somatostatinomas, PPomas, neurotensinomas. Semin. Oncol., 1987; 14: 263-281
[PubMed]
[38] Vinik A.I., Thompson N., Eckhauser F., Moattari A.R.: Clinical features of carcinoid syndrome and the use of somatostatin analogue in its management. Acta Oncol., 1989; 28: 389-402
[PubMed]
[39] Waldherr C., Pless M., Maecke H.R., Schumacher T., Crazzolara A., Nitsche E.U., Haldemann A., Mueller-Brand J.: Tumor response and clinical benefit in neuroendocrine tumors after 7.4 GBq (90)Y-DOTATOC. J. Nucl. Med., 2002; 43: 610-616
[PubMed]
[40] Wang C.: Mangafodipir trisodium (MnDPDP)-enhanced magnetic resonance imaging of the liver and pancreas. Acta Radiol. Suppl., 1998; 415: 1-31
[PubMed]
[41] Welin S.V., Janson E.T., Sundin A., Stridsberg M., Lavenius E, Granberg D., Skogseid B., Oberg K.E., Eriksson B.K.: High-dose treatment with a long-acting somatostatin analogue in patients with advanced midgut carcinoid tumours. Eur. J. Endocrinol., 2004; 151: 107-112
[PubMed] [Full Text PDF]
[42] Wymenga A.N.M., Eriksson B., Salmela P.I., Jacobsen M.B., Van Cutsem E.J.D.G., Fiasse R.H., Valimaki M.J., Renstrup J., de Vries E.G.E., Oberg K.E.: Efficacy and safety of prolonged-release lanreotide in patients with gastrointestinal neuroendocrine tumors and hormone related symptoms. J. Clin. Oncol., 1999; 17: 1111-1117
[PubMed] [Full Text HTML] [Full Text PDF]