
Negatywna regulacja sygnalizacji receptorów Toll-podobnych
Halina Antosz 1 , Dorota Choroszyńska 1Streszczenie
Mechanizm odporności wrodzonej działa w oparciu o receptory PRR – rozpoznające tzw. wzorce molekularne związane z patogenami – PAMPs. Wśród receptorów PRR wyróżnia się: receptory Toll-podobne (TLR). W wyniku kontaktu z patogenami, receptory TLR aktywują wewnątrz komórki swoiste szlaki sygnalizacyjne. Dzieje się to za pośrednictwem białek adaptorowych, tj. MyD88, TIRAP, TRIF, TRAM oraz IPS-1, które uczestniczą w kaskadowej aktywacji, kinaz: IKK, MAP, RIP-1, TBK-1 oraz czynników transkrypcyjnych (NF-κB, AP-1) i regulatorowych (IRF3). Następstwem aktywacji są cytokiny prozapalne, chemokiny, interferony i enzymy. Drogi aktywacji PRR są kontrolowane przez zewnątrz- i wewnątrzkomórkowe cząsteczki zapobiegające przed nadmierną ekspresją PRR. Są wśród nich rozpuszczalne postaci receptorów (sTLR), białka przezbłonowe (ST2, SIGIRR, RP105, TRAIL-R) oraz inhibitory wewnątrzkomórkowe (SOCS-1, SOCS-3 sMyD88, TOLLIP, IRAK-M, SARM, A20, β-arestyna, CYLD, SHP). Cząsteczki te zapewniają utrzymanie równowagi między aktywacją i hamowaniem oraz równoważenie korzystnych i niekorzystnych skutków rozpoznania antygenu.
Słowa kluczowe:NOD2 • SOCS • ST2L • SIGIRR • ß-arestyna • RP105 • TRAIL • sMyD88 • A20 • SHP • CYLD • SARM • Tollip • IRAK-M
Summary
The mechanism of innate immunity is based on the pattern recognition receptors (PRR) that recognize molecular patterns associated with pathogens (PAMPs). Among PRR receptors Toll-like receptors (TLR) are distinguished. As a result of contact with pathogens, TLRs activate specific intracellular signaling pathways. It happens through proteins such as adaptor molecules, e.g. MyD88, TIRAP, TRIF, TRAM, and IPS-1, which participate in the cascade activation of kinases (IKK, MAP, RIP-1, TBK-1) as well as transcription factors (NF-κB, AP-1) and regulatory factor (IRF3). The result of this activation is the production of active proinflammatory cytokines, chemokines, interferons and enzymes. The PRR pathways are controlled by extra – and intracellular molecules to prevent overexpression of PRR. They include soluble receptors (sTLR), transmembrane proteins (ST2, SIGIRR, RP105, TRAIL-R) and intracellular inhibitors (SOCS-1, SOCS-3, sMyD88, TOLLIP, IRAK-M, SARM, A20, β-arrestin, CYLD, SHP). These molecules maintain the balance between activation and inhibition and ensure balancing of the beneficial and adverse effects of antigen recognition.
Key words:NOD2 • SOCS • ST2L • SIGIRR • ß-arrestin • RP105 • TRAIL • sMyD88 • A20 • SHP • CYLD • SARM • Tollip • IRAK-M
Wprowadzenie
odporności: odporność nieswoista (wrodzona) oraz odporność swoista (nabyta). Układ nieswoisty reaguje nieswoiście, ale bardzo szybko. Jego pierwotnym zadaniem jest możliwość rozróżnienia potencjalnych antygenów obcych od własnych oraz identyfikacja fizjologicznej flory organizmu. Mechanizm ten działa w oparciu o istnienie określonej i ograniczonej liczby receptorów PRR (pattern recognition receptors), które selektywnie rozpoznają charakterystyczne ligandy, tzw. wzorce molekularne związane z patogenami – PAMPs (patogen associated molecular patterns) [1]. W komórkach odpowiedzialnych za odporność wrodzoną i nabytą, receptory PRR są umiejscowione na błonach komórkowych lub wewnątrz pęcherzyków cytoplazmatycznych. Wśród PRR wyróżnia się trzy rodzaje receptorów: Toll-podobne (TLR) rozpoznające pierwotniaki, grzyby, bakterie i wirusy, receptory NLR (NOD-like receptors) rozpoznające tylko bakterie oraz receptory RLR (RIG-I-like receptors) rozpoznające wyłącznie wirusy.
Receptory Toll-podobne
Dotychczas u ludzi zidentyfikowano 10 typów receptorów Toll-podobnych. TLR1, TLR2, TLR4, TLR5, TLR6, TLR10 zlokalizowano na błonie komórkowej zaś TLR3, TLR7, TLR8 i TLR9 w błonach pęcherzyków wewnątrzcytoplazmatycznych w obrębie endosomów i lizosomów [48]. Ze względu na lokalizację, receptory TLR uczestniczą w indukcji odpowiedzi immunologicznej w chwili inwazji patogenu. Są ogniwem łączącym odporność nieswoistą ze swoistą. Indukcja odpowiedzi swoistej jest następstwem aktywacji TLR komórek prezentujących antygeny, które intensyfikują te procesy. Receptory TLR są przezbłonowymi glikoproteidami. N-końcowa domena cząsteczki TLR, tzw. LRR (leucine rich repeats) zbudowana jest z tandemowych powtórzeń bogatych w leucynę, C-końcowa domena, cytoplazmatyczna TIR (Toll-IL-1-receptor), tak zwana ze względu na dużą homologię do receptora interleukiny 1 typu 1 (IL-1R1). Domena TIR jest najważniejsza w uruchomianiu kaskady sygnałów, w następstwie kontaktu z cząsteczkami adaptorowymi. Białkami adaptorowymi są cząsteczki TIRAP, TRIF, TRAM i MyD88 [49,121]. Są one selektywnie angażowane przez receptory TLR. Oprócz TLR3 wszystkie szlaki sygnalizacyjne wymagają białka MyD88. Wyróżnia się więc szlaki MyD88-zależne i MyD88-niezależne [94]. Pobudzone receptory TLR5, TLR7, TLR9 łączą się z MyD88 bezpośrednio [56]. Do aktywacji TLR2 i TLR4, niezbędne jest białko adaptorowe TIRAP [22,56]. Jest to związane z obecnością jednoimiennych ładunków elektrycznych receptorów TLR2, TLR4 i cząsteczki adaptorowej MyD88 [22]. Powierzchnia receptorów TLR2, TLR4 i MyD88 jest elektrododatnia, przez co niemożliwe jest bezpośrednie przyłączenie MyD88 do tych receptorów. Cząsteczka TIRAP ma natomiast powierzchnię głównie elektroujemną, co pozwala na wiązanie się do niej elektrododatnich cząsteczek TLR2, TRLR4, MyD88 i tworzenie kompleksu sygnalizacyjnego [42]. Ponadto TIRAP zawiera domenę odpowiedzialną za wiązanie PIP2 (fosfatydyloinozytolo-4,5-bisfosforan). PIP2 to dyskretny, lecz istotny region błony plazmatycznej, nadaje zdolność białku TIRAP rekrutacji MyD88 do błony komórkowej [5]. O istocie tego połączenia świadczą obserwacje, z których wynika, że mutacje TIRAP w domenie wiążącej PIP2 lub hydroliza komórkowego PIP2 przez bakteryjną fosfatazę PIP2, znosi możliwość przyłączania się TIRAP do błony plazmatycznej i zdolności TIRAP do indukcji cytokin w odpowiedzi na LPS [23].
Na skutek bezpośredniego lub pośredniego przyłączenia cząsteczki adaptorowej MyD88 do TLR, następstwem jest pobudzenie tych receptorów i aktywacja kinazy IRAK-4. Etap ten prowadzi z kolei do fosforylacji IRAK-1. Aktywna kinaza IRAK-1 zostaje uwolniona do cytoplazmy, gdzie łączy się z czynnikiem TRAF6, powodując aktywację kompleksu TAK1/TAB [49]. Pobudzony kompleks TAK1/ TAB powoduje aktywację kinazy czynnika IκB (IKK – IκB kinase) oraz kinazy MAP (mitogen-activated protein). Aktywacja kinazy IKK jest związana z fosforylacją i degradacją inhibitora IκB (inhibitor czynnika jądrowego NF-κB), czego następstwem jest uwolnienie czynnika transkrypcyjnego NF-κB (nuclear factor-κB). NF-κB wnika do jądra komórkowego i indukuje ekspresję wielu genów, w tym kodujących cytokiny prozapalne, cząsteczki adhezyjne i liczne enzymy [121].
Transdukcja sygnału od receptora TLR3, przebiega bez udziału MyD88. Kaskada sygnałowa uruchamiana jest za pośrednictwem cząsteczki adaptorowej TRIF (TIR-domain-conatining adaptor protein including IFN-beta). TRIF oddziałuje z cząsteczkami TRAF3 i TRAF6 (tumor necrosis factor – TNF receptor-associated factor), powodując aktywację dwóch równoległych szlaków, których końcowym efektem jest również aktywacja genów zależnych od NF-κB (ryc. 1).

Ryc. 1. Szlaki wewnątrzkomórkowej transdukcji sygnałów od receptorów TLR, RLR i NRL (wg [54] zmodyfikowano)
Receptory RLR
Receptory RLRs są wewnątrzkomórkowymi białkami pełniącymi funkcję receptorów zdolnych do wykrywania wirusowego RNA w cytoplazmie [67,69]. Zidentyfikowano trzy RLRs, tj. RIG-I (retinoic acide inducible gene-1) znany również jako DDX58, MAD5 (melanoma differentiation-associated gene) znany również jako Helicard i trzeci receptor LGP2. Cząsteczki białkowe RIG-I i MDA5 na N-końcu zawierają domenę aktywacji i rekrutacji kaspaz (CARD – caspase activation and recruitment domain) oraz domenę RNA helikazy. Na C-końcu znajduje się domena regulatorowa lub domena represorowa (CTD – carboxy-terminal domain) [75]. Domeny helikazy i CTD są odpowiedzialne za wiązanie wirusowego RNA, zaś domena CARD jest wymagana do sygnalizacji [117]. Domena LGP2 jest homologiem RIG-I i MAD5, brakuje jej jednak domeny CARD i tym samym nie ma możliwości sygnalizacji [88,118]. Ekspresja LGP2 jest indukowana po traktowaniu dsRNA (double-stranded RNA), IFN albo przez zakażenie wirusem [85].
RIG-I rozpoznaje 5’trifosforan ssRNA (single-stranded RNA) oraz krótkie dsRNA. Aktywacja RIG-I w odpowiedzi na infekcję wirusową lub syntetyczny RNA wprowadzony do cytoplazmy generuje w odpowiedzi INF (interferon) typu I [118].
Receptor MAD5 odpowiedzialny jest za detekcję wirusów Picornaviridae [106], struktury dsRNA i stosunkowo długie struktury (>1 κB) poly I:C (polyinosinic:polycytidylic acid – struktury podobne do wirusowego dsRNA). Krótkie struktury (<1 κB) poly I:C rozpoznawane są przez RIG-I [96].
Cząsteczka RIG-I jest połączona ze szlakami sygnałowymi, aktywującymi czynniki NF-κB, MAPKs i IRFs (interferon regulatory factors), które przyczyniają się do wydzielania białka adaptorowego IPS-1 (interferon-beta promoter stimulator 1). N-koniec IPS-1 zawiera domenę podobną do CARD, wykazując homologię z RIG-1, natomiast domena C-końcowa zawiera segment transbłonowy, którego celem są mitochondria [43,68,91,112]. Do N-końca IPS-1 przyłącza się białko TRAF3 [92], które wykazuje aktywność ligazy E3 ubikwityny. TRAF3 ulega ubikwitylacji co prowadzi do aktywacji kinaz TBK1 (TANK-binding kinase 1; TANK-TRAF-associated NF-kappa B activator) i IKKi (inducible IκB kinase), które następnie fosforylują czynniki transkrypcyjne IRF-3 i IRF-7. W jądrze czynniki te aktywują ekspresję genu interferonu typu I.
IPS-1 może aktywować również inny szlak. Poprzez kaskadę białek FADD (Fas-associated death domain-containg protein), kaspazę 8 i kaspazę10 aktywacji ulega czynnik NF-κB, odpowiedzialny za indukcję cytokin prozapalnych [97].
Receptory NOD-podobne – NLR
Receptory NLR (nucleotide binding domain leucine-rich repeat) rozpoznają wewnątrzkomórkowe struktury bakteryjne PAMP [64]. Do NLR należą białka NOD1 i NOD2 (nucleotide-binding oligomerization proteins), które są zróżnicowane ze względu na ich specyfikę wobec liganda. Ligandem dla NOD1 jest kwas γ-D-glutamylo-mezo-diaminopimelinowy (iEDAP) [14], charakterystyczny dla bakterii Gram-ujemnych i pewnych bakterii Gram-dodatnich. Rola NOD2 polega na rozpoznawaniu minimalnej struktury, jaką jest dipeptyd muramylowy (MDP) stanowiący część peptydoglikanu bakteryjnej ściany komórkowej [70]. Przyłączenie ligandów do NOD1 i NOD2 prowadzi do uruchomienia kaskady sygnalizacyjnej i aktywacji kinaz p38 MAP, ERK (extracellular signal-regulated protein kinase) i JNK (c-Jun N-terminal kinase) oraz NF-κB, co skutkuje wytwarzaniem cytokin [80]. Stymulacja NOD1 i NOD2 indukuje rekrutację białka RIP2 (receptor-interacting protein 2; znanego także jako Rick) i CARD9, które wspierają szlaki sygnalizacji aktywowane przez MAPK i zależną od NF-κB aktywację genów [79]. RIP2 aktywuje IKK, bezpośrednio lub przez rekrutację TAK1, prowadząc do fosforylacji i degradacji IκB. Następstwem jest uwolnienie NF-κB i wytwarzanie cytokin prozapalnych i chemokin. Szlak MAPK jest również aktywowany przez RIP2 i TAK1, choć szczegóły tego szlaku pozostają do wyjaśnienia [79,109].
NOD2 (znany również jako CARD 15) jest jednym z 22 cytoplazmatycznych białek zidentyfikowanych u ludzi, należącym do grupy receptorów NLR. Niektóre wyniki badań sugerują, że NOD2 może działać jako inhibitor sygnalizacji TLR2 [114]. Badania u ludzi, jak dotąd nie wykazały negatywnej regulacji interakcji między NOD2 i TLR2, a w rzeczywistości wskazują na efekt synergiczny między tymi cząsteczkami [7].
Regulatory negatywne
Sposób aktywacji ze wszystkich PRR jest kontrolowany przez wiele zewnątrz- i wewnątrzkomórkowych cząsteczek, utrzymujących równowagę między aktywacją i hamowaniem oraz równoważeniem korzystnych i niekorzystnych skutków rozpoznania antygenu.
Mechanizm negatywnej regulacji z TLR najlepiej rozwinięty jest w komórkach immunologicznych. Jest on ściśle ograniczony do konkretnego TLR i funkcjonuje na wielu poziomach sygnalizacji TLR. Blokowanie sygnałów z receptorów Toll-podobnych rozpoczyna się już na powierzchni komórek, a swój udział w tym procesie mają rozpuszczalne postaci receptorów TLR (sTLR – soluble TLR), sTLR4, sTLR2 oraz sST2. Następnie aktywność inhibicyjną przejmuje rodzina białek przezbłonowych z domeną TIR. Należą do niej białka ST2, SIGIRR, RP105, TRAIL-R. Kolejne to inhibitory cytoplazmatyczne kontrolujące funkcje białek adaptorowych i kinaz zaangażowanych w wewnątrzkomórkowy przekaz sygnału. Są to białka MyD88s, A20, CYLD, β-aresyna, SARM, SOCS, TOLLIP, IRAK-M, które hamują sygnały na różnych etapach aktywacji (ryc. 2).
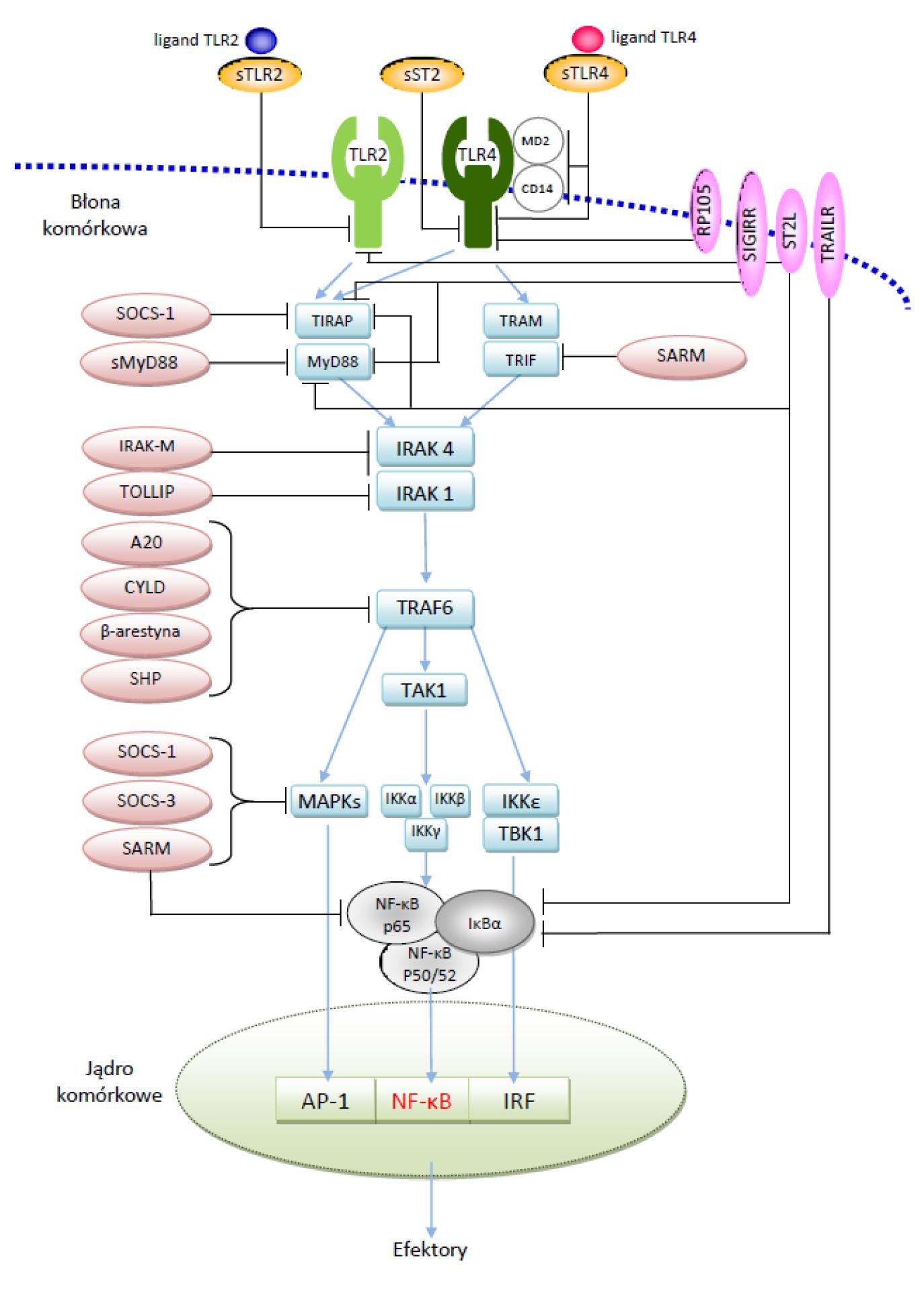
Ryc. 2. Negatywna regulacja sygnalizacji z TLR (opracowanie autorskie)
Negatywne regulatory zewnątrzkomórkowe
sTLR
U ssaków, w tym u ludzi, wykryto różne izoformy produktów genów TLR2 i TLR4, powstałe na skutek potrans-lacyjnej modyfikacji białek [51]. Dotąd zidentyfikowano co najmniej dwie rozpuszczalne postaci TLR receptorów (sTLRs). Te naturalnie wytwarzane cząsteczki służą jako pierwsza linia negatywnej regulacji, zapobiegająca przed nadmierną aktywacją gospodarza, po infekcji drobnoustrojami. sTLR4 i sTLR2 mogą hamować sygnalizację przez blokowanie interakcji między TLR4 lub TLR2 i innych kooperantów receptorów, zwłaszcza MD2 i CD14. Badania in vitro wykazały, że w makrofagach indukowanych LPS, zrekombinowana postać sTLR4 hamuje aktywację NF-αB i TNF-α, natomiast w monocytach stymulowanych agonistą TLR2 – lipopeptydem bakteryjnym, sTLR2 hamuje IL-8 i wytwarza TNF-α [36].
ST2 (suppression of tumorigenicity 2)
U ludzi gen ST2 (locus 11p14.3-p12), jest członkiem rodziny receptora IL-1. Koduje dwa główne produkty. W wyniku alternatywnego składania transkryptu powstają dwie izoformy ST2L i sST2. ST2L jest silnie glikozylowanym białkiem błonowym (61,5-80 kD), z zewnątrz- i wewnątrzkomórkowymi domenami. W regionie zewnątrzkomórkowym zawiera trzy domeny immunoglobulinowe, częścią wewnątrzkomórkową jest domena TIR.
Izoforma sST2 jest rozpuszczalnym białkiem wydzielanym poza komórkę, które składa się tylko z części zewnątrzkomórkowej cząsteczki ST2L.Yin i wsp. [116] stwierdzili, że sST2 funkcjonuje jako negatywny regulator TLR4. W badaniach in vitro wykazano, że sST2 może hamować wytwarzanie cytokin prozapalnych przez makrofagi stymulowane LPS. sST2 jest wydzielane przez aktywowane komórki Th2, które wykazują na swej powierzchni cząsteczkę ST2L. Sugeruje się, że ta skrócona izoforma odgrywa ważną rolę w regulacji biologicznej aktywności IL-33, przez działanie jako receptor „przynęta”, zapobiegając interakcji IL-33 z ST2L [52].
Negatywne regulatory błonowe
ST2L
Badania wykazały, że ST2 negatywnie reguluje sygnalizację TLR4 tłumiąc wytwarzanie cytokin prozapalnych indukowanych LPS. Mechanizm działania polega na zahamowaniu sygnalizacji białek adaptorowych MyD88 i TIRAP, albo poprzez związanie ich domen TIR [8], albo poprzez hamowanie degradacji IκBα. Następstwem jest brak czynnika transkrypcyjnego NF-κB dla promotora cytokin prozapalnych IL-6, IL-12 i TNF-α, [95,98]. Niedawne badania Liu i wsp. [58] wykazały, że ST2L jest negatywnym regulatorem również TLR2, a mechanizm hamowania, podobnie jak w przypadku TLR4, związany jest z inhibicją białek adaptorowych MyD88 i TIRAP.
SIGIRR (single-immunoglobulin and toll – interleukin1 receptor)
Gen SIGIRR (locus 11p15.5) koduje białko zaliczane do rodziny IL-1R. Jego zewnątrzkomórkowa część zawiera pojedynczą domenę immunoglobulinową. Ze względu na obecność domeny TIR białko to znane jest również jako TIR8. SIGIRR, w przeciwieństwie do większości opisanych inhibitorów TLR, wykazuje bardzo swoisty wzór ekspresji. U ludzi jest on ograniczony tylko do monocytów (ale nie makrofagów) i niedojrzałych komórek dendrytycznych (DC) [111]. Funkcjonuje jako inhibitor receptorów IL-1R, TLR-4, ale mechanizm hamowania w obu przypadkach jest inny. Obie domeny SIGIRR, zewnątrzkomórkowa Ig i wewnątrzkomórkowa TIR, są niezbędne do hamowania sygnalizacji IL-1 natomiast tylko domena TIR jest konieczna do hamowania sygnalizacji LPS. Niemniej jednak konsekwencją tej inhibicji jest blokowanie funkcji białek adaptorowych MyD88 i TRAF6 [84]. Potwierdziły to badania komórek dendrytycznych z niedoborem SIGIRR, które wykazywały zwiększone wytwarzanie cytokin w odpowiedzi na ligandy TLR, tj LPS i CpG DNA. SIGIRR wpływa na sygnalizację z TLR4 hamując zarówno MyD88-zależną, jak i MyD88-niezależną ścieżkę [41,88].
SIGIRR prawdopodobnie bezpośrednio włącza się w powstanie kompleksu receptora, w tym w dimeryzację receptora, właściwą rekrutację i aktywację komponentów sygnalizacyjnych, hamując dalsze zdarzenia. Oprócz bezpośredniego hamowania kompleksów receptora, SIGIRR może aktywnie rekrutować wewnątrzkomórkowe cząsteczki hamujące receptory, przez co działa hamująco [91]. Wykazano, że SIGIRR hamuje syntezę IL-1α i IL-1, czego efektem jest osłabione powstawanie komórek pomocniczych Th17 (wydzielających IL-17) [93].
RP105 (CD180)
Produktem genu RP105 (locus 5q12) jest powierzchniowa cząsteczka białkowa bogata w powtórzenia leucynowe, której zewnątrzkomórkowa domena wykazuje homologię receptora LPS, aczkolwiek jego ekspresja, w przeciwieństwie do TLR4, ograniczona jest wyłącznie do komórek prezentujących antygen (komórki B, makrofagi i komórki dendrytyczne) [19,72]. Doniesienia na temat białka RP105 są kontrowersyjne, ponieważ przypisuje się mu przeciwstawne funkcje. CD180, opisywany jest zarówno jako wymagany koreceptor dla LPS i stymulator limfocytów B [73] oraz jako inhibitor TLR4 w komórkach DC, bez fizjologicznego wpływu na limfocyty B [20].
TRAIL (tumor necrosis factor (TNF)-related apoptosis inducing ligand)
Cytokina TRAIL była zidentyfikowana jako trzeci członek nadrodziny TNF, który indukuje apoptozę w wielu nowotworowych liniach komórkowych [83,108]. Zainteresowanie tą nową cytokiną wzrosło dodatkowo po odkryciu, że TRAIL może zabijać komórki nowotworowe in vivo nie wywołując toksyczności [102]. Badania wykazały, że poza indukcją apoptozy, kompleks TRAIL/ TRAIL-receptor odgrywa rolę w regulacji homeostazy układu odpornościowego oraz w nadzorze immunologicznym nowotworów.
TRAIL wykazuje ekspresję zarówno jako transmembranowe białko typu II, z zewnątrzkomórkowym C-końcem oraz jako rozpuszczalny trimer. TRAIL wiąże się ze złożonym systemem receptorów z różnym powinowactwem [89]. U ludzi poznano receptory: TRAIL-R1 (DR4) oraz TRAIL-R2 (DR5/APO2) z wewnątrzkomórkowym konserwatywnym motywem zwanym domeną śmierci (DD), która jest warunkiem indukcji apoptozy, TRAIL-R3 (DcR 1, TRID), który nie ma cytoplazmatycznej domeny DD oraz TRAIL-R4 (DcR2, TRUNDD), który ma skróconą domenę DD. Dwie ostatnie postaci receptorów nie mogą przekazywać sygnałów apoptotycznych, ale uważa się, że funkcjonują jako „przynęty” białek, które mogą hamować sygnalizację za pośrednictwem TRAIL-R1 i TRAIL-R2 [2].
Badania na myszach wykazały, że ligandy TLR2, TLR3, TLR4, zwiększają ekspresję TRAIL-R. Natomiast w przypadku deficytu TRAIL-R, w odpowiedzi na te ligandy, widoczne jest zwiększone wytwarzanie cytokin. Diehl i wsp. [17] twierdzą, że TRAIL-R hamuje sygnalizację TLR poprzez stabilizowanie IκBα, w wyniku czego następuje obniżenie ekspresji, jednego z najistotniejszych w odpowiedzi odpornościowej, czynnika transkrypcyjnego – NF-κB. Te dane sugerują, że TRAIL-R jest ważnym negatywnym regulatorem cytokin we wrodzonym układzie odpornościowym.
Negatywne regulatory wewna¸trzkomórkowe
MyD88s
Cząsteczka MyD88 jest najbardziej uniwersalnym białkiem adaptorowym w sygnalizacji TLR. Jej skrócona postać MyD88s, różni się od postaci dzikiej, brakiem domeny ID (intermediate domain), pośredniczącej w integracji z IRAK-4. Z dostępnych danych wynika, że ekspresja MyD88s ograniczona jest do komórek śledziony i w niewielkim stopniu do neuronów mózgu [40]. W badaniach in vitro stwierdzono zwiększoną ekspresję MyD88s po 16-godzinnej stymulacji LPS, w komórkach ludzkiej linii monocytarnej (THP-1) [38]. Następnie Burns i wsp. [12] wykazali, że zwiększona ekspresja MyD88s prowadzi do utworzenia heterodimeru MyD88s-MyD88 zamiast homodimeru MyD88-MyD88 wymaganego do rekrutacji IRAK-4. W tych warunkach kinaza IRAK-1 jest angażowana przez swoją domenę DD do interakcji z MyD88s, ale nie jest fosforylowana przez IRAK-4, ponieważ MyD88s nie może się łączyć z IRAK-4. Na tym etapie sygnalizacja TLR zostaje zahamowana. Oznacza to, że cząsteczka MyD88s może być zaangażowana w ujemny mechanizm regulacyjny sprzężenia zwrotnego, który kontroluje nadmierną aktywację TLR [39].
A20 (TNFAIP3 – tumor necrosis factor-induced protein 3)
Ekspresja A20, indukowana TNF-α, jest szybka i przemijająca, z maksymalną aktywnością po 1 godzinie [21]. W małych ilościach A20 występuje w tkankach niestymulowanych, ale po infekcji bakteryjnej lub wirusowej ekspresja wzrasta. Wykazano, że białko to reguluje sygnalizację TLR, zainicjowaną przez komensale bakteryjne. W sytuacji braku A20, zainicjowana sygnalizacja, nie może być obniżana, co prowadzi do załamania tolerancji wrodzonego układu odpornościowego w stosunku do własnej mikroflory jelitowej [99,100].
Cząsteczka A20 w swej strukturze zawiera 790-aminokwasów. Jej N-końcowa domena OTU (ovarian tumor) – charakterystyczna jest dla nadrodziny proteaz cysteinowych, C-koniec natomiast ma strukturę palców cynkowych [107]. Domena OTU zawiera, powiązane z ubikwityną i oddziałujące na siebie motywy wspólne dla enzymów deubikwitynacji (DUBs) [4], jednak A20 nie pełni na ogół funkcji DUB [57]. C-końcowa domena składa się z siedmiu palców cynkowych. Sześciu o sekwencji Cys-X4-Cys-X11-Cys-X2-Cys i jednego motywu ze strukturą Cys-X2-Cys-X11-Cys-X2-Cys. Taka część budowy nie jest znana w innych białkach i w związku z tym A20 tworzy nową klasę białek o strukturze palców cynkowych [78]. Aktywność ligazy ubikwityny zawiera czwarty palec cynkowy [101], pozostała część C-końca odpowiedzialna jest za integrację z lizosomami i degradację cząsteczek sygnałowych [54].
Białko A20 jest negatywnym regulatorem, który może kontrolować zarówno MyD88 zależne i niezależne od MyD88 szlaki sygnalizacyjne TLR. Zaangażowane jest w aktywację NF-κB na szlaku receptorów TLR2 i TLR4 [6], gdzie hamuje białko TRAF6 współdziałające z kinazą IRAK-1 w stymulacji kompleksu TAK1/TAB i dalej w aktywacji kinaz IKKα i IKKβ inhibitora IκB. Brak fosforylacji IκB stabilizuje kompleks IκB/NF-κB w cytoplazmie i nie dochodzi w jądrze komórkowym do aktywacji istotnych genów przez czynnik transkrypcyjny NF-κB. Badania in vitro na myszach pozwoliły na sformułowanie wniosku, że wskutek stymulacji LPS, oś A20-TRAF6 jest głównym elementem, który zapobiega aktywacji NF-κB. Wyciszenie aktywności A20 pozwala na przywrócenie funkcji TRAF6 i aktywację NF-κB po stymulacji LPS [61].
Blokowanie aktywacji NF-κB przez A20 odbywa się też w sposób zależny od TNF-α. Wiązanie TNF-α do jego receptora powoduje rekrutację domen DD TNF-receptora, które następnie współdziałąją z kinazą serynowo-treoninową RIP (receptor-interacting protein) albo z TRAF2 (TNF-receptor associated factor 2). Poprzez ingerowanie w sygnalizację RIP i TRAF2, A20 blokuje w tym miejscu aktywację szlaku NF-κB [30]. Nie dochodzi do aktywacji kinazy IKK i dalej do degradacji inhibitora IκB. Czynnik NF-κB pozostaje zablokowany w cytoplazmie.
CYLD (cylindromatosis)
CYLD jest kolejnym negatywnym regulatorem wrodzonej odpowiedzi przeciwwirusowej regulowanej przez RIG-I (retinoic acid-inducible gene 1) [24]. Z badań Yoshida i wsp. [119] wynika, że ligandy TLR2 indukują aktywację kinaz IKK-IκBα oraz MKK3/6-p38 nie tylko przez TRAF6, ale również przez TRAF7 Aktywacja obu kinaz może indukować transkrypcję TNF-α, IL-1β i IL-8 oraz CYLD. CYLD z kolei prowadzi do zahamowania TRAF6 i TRAF7 prawdopodobnie w sposób zależny od deubikwitynacji. Ponadto wykazano, że CYLD negatywnie reguluje również aktywację JNK indukowaną TNF-α.
Biorąc pod uwagę, że poliubikwitynacja odgrywa istotną rolę w aktywizacji różnych szlaków sygnałowych, jest prawdopodobne, że CYLD może negatywnie regulować więcej dróg sygnalizacyjnych.
ß-arestyna
U ssaków rodzina arestyn składa się z czterech izoform. Dwie z nich występują swoiście w fotoreceptorach siatkówki oka, natomiast dwie pozostałe, tj. β-arestyna 1, β-arestyna 2 są powszechnie występującymi białkami cytosolowymi. Podstawową funkcją wszystkich arestyn jest zdolność blokowania przekazywania sygnału wewnątrzkomórkowego przez aktywowane receptory sprzężone z białkami G [27]. Ponadto β-arestyna, przez obecność domeny wiążącej klatryny, pośredniczy w endocytozie i recyklizacji receptorów [65] oraz uczestniczy w sygnalizacji wewnątrzkomórkowej wchodząc w interakcje, m.in. z kinazami białkowymi rodziny Src [60] i MAPK [53]. Poprzez oddziaływania z fosfodiesterazami może modulować poziom cAMP w komórce [82]. Badania Witherowa i wsp. [110] wykazały, że β-arestyna jest zaangażowana w modulowanie sygnalizacji z TLR poprzez wpływ na aktywność NF-κB. Zaobserwowano, że zarówno zwiększona ekspresja β-arestyny1 i β-arestyny 2 skutkuje obniżeniem aktywności NF-kB. Jednocześnie stwierdzono, że β-arestyna 1 hamuje NF-κB znacznie silniej niż β-arestyna 2. Badania Wanga i wsp. [104] na myszach, pozwoliły precyzyjnie zlokalizować miejsce inhibicji na ścieżce sygnalizacyjnej TLR. Okazało się, że po aktywacji domeny TIR-TLR, β-arestyna1 oddziałuje z TRAF6. Utworzony kompleks zapobiega autoubikwitynacji i oligomeryzacji TRAF6 wymaganej do aktywacji NF-κB i AP-1. Stwierdzono ponadto, że myszy z deficytem β-arestyny traktowane endotoksyną wykazują wysoką ekspresję cytokin prozapalnych i są bardziej podatne na szok endotoksyczny, co sugeruje, że β-arestyny są niezbędnym negatywnym regulatorem odporności wrodzonej regulowanej sygnałami TIR.
SARM (sterile α– Armadillo-motif)
Gen SARM (locus 17q11) koduje 690-animokwasowe białko SARM zawierające N-końcową domenę ARM (Armadillo motif), domenę SAM (sterile α motif) i C-końcową domenę TIR [71,76]. Domena N-końcowa zawiera wielozasadowy motyw i region bogaty w glicynę (GRR), co prawdopodobnie wpływa na przestrzenną lokalizację i aktywację SARM [81]. Domena TIR jest najlepiej zachowaną domeną, po Drosophila [71] i Caenorhabditis elegans [15]. Unikatowa kombinacja tych trzech domen w SARM sprawia, że białko to działa inaczej niż cząsteczki adaptorowe [66,76]. Funkcja SARM jest stosunkowo mało znana, ale wydaje się odgrywać wiele ról, które są odmienne w różnych gatunkach, tkankach i okolicznościach. Podczas infekcji SARM jest umiejscowiony w cytoplazmie i oddziałuje bezpośrednio na cytosolowy TRIF, co powoduje obniżenie aktywności NF-κB, IRF3 (interferon-regulatory factor3) sygnalizacji TRIF-zależnej, indukowanej z TLR3 i TLR4 [13].
Z najnowszych doniesień Penga i wsp. [81] wynika, że u ludzi SARM blokuje nie tylko TRIF – i MyD88-zależną, indukowaną aktywację czynnika transkrypcyjnego AP-1, ale również endogenną postać AP-1. Stąd sugeruje się, że SARM może również odgrywać rolę w autoimmunizacji.
Regulacja ekspresji SARM zachodzi zarówno na poziomie mRNA jak i białka. Odnotowano, że w godzinę po stymulacji LPS, transkrypcja SARM gwałtownie nasila się, ale w przeciągu 6 godzin jest stłumiona. Zwiększona ekspresja SARM powoduje zahamowanie fosforylacji p38 MAPK, co dowodzi, że SARM pokonuje nadmierną reakcję odpornościową poprzez wyłączenie aktywności MAPK w celu regulacji sygnalizacji immunologicznej [81].
SHP (small heterodimer partner)
SHP znany jest jako sierocy receptor jądrowy (orphan nuclear receptor), który uczestniczy w regulacji transkrypcji różnych szlaków metabolicznych. Według Yuki i Jo [121] jest również negatywnym regulatorem sygnalizacji TLR. Badania szpikowej linii komórkowej z niedoborem SHP wykazały, że w odpowiedzi na różne ligandy TLR, komórki te wydzielały duże ilości cytokin prozapalnych i chemokin w porównaniu z komórkami typu dzikiego. Inne badania dowiodły, że w komórkach spoczynkowych SHP hamuje sygnalizację zależną od NF-κB przez oddziaływanie z p65-NF-κB. Natomiast po stymulacji SHP osłabia poliubikwitynację TRAF6, związaną z K63 (miejsce przyłączenia ubikwityny do Lys-63), poprzez oddziaływanie z domeną RING TRAF6 [122].
SOCS1
SOCS1 (suppressor of cytokine signalling) jest jednym z ośmiu członków rodziny białek SOCS syntetyzowanych w wyniku wczesnej odpowiedzi genów na wiele bodźców zewnętrznych, tj. cytokiny, czynniki wzrostu, ligandy receptorów Toll, statyny i cykliczny adenozyno-monofosforan (cAMP) [32,35].
Regulacja odpowiedzi immunologicznej z udziałem białek SOCS jest związana z budową oraz występowaniem w ich strukturze charakterystycznych domen funkcjonalnych, tj.: domeny N-końcej o różnej sekwencji i długości aminokwasów zakończonej regionem KIR (kinase inhibitor region) [125], domeny centralnej SH2 (Src homology 2 protein) oraz tzw. motywu SOCS box, tj. domeny C-końcowej [18]. N-końcowa domena KIR jest niezbędna do zahamowania kinazy JAK2 [47], po indukcji cytokinami zapalnymi IL-2, IL-3, IL-6, IFN-γ i erytropoetyną. Centralna domena SH2 wiąże różne motywy fosforylowanych tyrozyn białek docelowych, w tym aktywowanych receptorów cytokin [31]. N-koniec, regionu SH2 przylegający do sekwencji KIR jest rozszerzony o subdomenę N-ESS, która spełnia główne funkcje zarówno dla SOCS1 jak i SOCS3 [87,115]. Domena SOCS-box [44], jest niezbędna do zaktywowania sytemu ubikwityna/transferyna, ponieważ stanowi punkt przyłączenia dla ligaz biorących udział w procesie ubikwitynacji cząsteczek [18].
SOCS1 tłumi zarówno MyD88-zależną, jak i MyD88-niezależą ścieżkę sygnalizacji poprzez bezpośrednie działanie na elementy kaskady sygnalizacyjnej [45]. Kompleks TLR2-ligand, podobnie jak TLR4-ligand, rozpoznawany jest przez białko adaptorowe TIRAP/MAL, które pośredniczy w przekazywaniu sygnału, uruchamiając szlak MyD88 – zależny [33,114]. Mansell i wsp. [62] dowiedli, że TIRAP jest bezpośrednim celem białka SOCS1. Na skutek zwiększonej ekspresji SOCS1, TIRAP ulega poliubikwitynacji i degradacji, w konsekwencji dochodzi do zahamowania przekazu sygnału. Kolejnym etapem na tym szlaku jest fosforylacja IκBα przez kompleks IRAK-4 i IKK. Reakcja ta może również być hamowana przez białka SOCS1. Cząsteczki te poprzez domenę SH2 mogą tworzyć kompleks z kinazą IRAK-4. Utworzenie kompleksu prowadzi do degradacji IRAK-4 w proteasomie, przez co tłumiona jest fosforylacja IκBα przez IKK [74]. Ryo i wsp. [86] wykazali, że celem SOCS1 jest również podjednostka p65/RelA NFκB. SOCS1 poprzez ubikwitynację powoduje proteolizę cząsteczki NF-κB, co skutkuje obniżeniem aktywności tego transkrypcyjnego czynnika.
SOCS3
Białko SOCS3 to drugi najlepiej poznany członek rodziny SOCS. Jego funkcją jest regulacja następstw niepożądanych stymulacji LPS, przez modulowanie sygnałów wielu cytokin w tym IL-1, -10, -6 i TGF-β. Wszystkie te cytokiny wpływają na zwiększenie ekspresji SOCS3. Yoshimura i wsp. [120] wykazali, że zwiększona ekspresja białka SOCS3 hamuje TRAF6 na szlaku sygnalizacyjnym z receptora IL-1R. Represja aktywacji TRAF6 blokuje późniejsze włączenie białka TAK1, które jest wymagane do aktywacji zarówno ścieżki NF-κB jak i MAPK [25].
TOLLIP
Białko TOLLIP (Toll-interacting protein), jest endogennym modulatorem sygnalizacji receptorów Toll-podobnych, małą cząsteczką wiążącą ubikwitynę, odgrywającą rolę w kierowaniu białek do endosomów, należącą do grupy adaptorowych cząstek blokujących [77].
U ludzi gen transkrybowany jest w czterech różnych izoformach: TOLLIP.A, TOLLIP.B, TOLLIP.C, TOLLIP.D. Kanoniczna postać – TOLLIP.A, składa się z trzech funkcjonalnych domen: N-końcowej domeny TBD (Tom1 binding domain), centralnej domeny C2-podobnej i C-końcowej domeny CUE ( Coupling of ubiquitin to endoplasmic reticulum for degradation) [9]. Skrócone izoformy TOLIIP.B i D nie zawierają domeny TBD, zaś postać TOLLIP.C pozbawiona jest domeny C2 [59]. Rola izoform niekompletnych obecnie jest nieznana, aczkolwiek nie wyklucza się ich funkcji w sygnalizacji z TLR.
Domena TBD TOLLIP.A odpowiada za połączenie z cytoplazmatycznym białkiem Tom1 (Target of Myb1), przez to razem z ubikwityną i klatryną uczestniczy w sortowaniu białek we wczesnych endosomach [113].
Domena C2 selektywnie wiąże produkty kinazy PI3 (fosfoinzytolowa kinaza 3), tj. fosfatydyloinozytolo-3-fosforan [PtdIns(3)P] i fosfatydyloinozytolo-3,4,5-fosforan [PtdIns(3,4,5)P] [55], które są zaangażowane w wielu procesach sygnalizacyjnych [63]. Zakłócenie funkcji domeny C2 powoduje paraliż negatywnej regulacji, jaką wywiera TOLLIP na aktywację NF-κB, indukowaną LPS [55]. Domena CUE jest miejscem wiązania ubikwityny. Zakłócenie funkcji CUE skutkuje akumulacją IL1-RI w późnych endosomach [9].
W komórkach spoczynkowych, TOLLIP kontroluje aktywację ścieżki NF-κB zależnej od MyD88 na dwóch różnych poziomach. Po aktywacji LPS, TOLLIP w połączeniu z receptorami IL-1RI, TLR2 i TLR4, hamuje odpowiedź immunologiczną [10,124]. TOLLIP, jako inhibitor kinazy IRAK-1, wiąże ją uniemożliwiając jej autofosforylację [11] nie promując jednak degradacji IRAK-1 [55].
TOLLIP kontroluje wielkość wytwarzania cytokin prozapalnych w odpowiedzi na stymulację IL-1 i LPS. W przypadku braku infekcji białko to jest konieczne do utrzymywania komórek odpornościowych w spoczynku, natomiast podczas stanu zapalnego ułatwia wygaśnięcie sygnalizacji indukowanej z IL-1R i TLR poprzez zahamowanie aktywności IRAK-1. TOLLIP jest zaangażowany w tolerancji endotoksycznej [11,124], która jest czynnikiem adaptacyjnym, ograniczającym zakres i czas trwania produkcji cytokin zapalnych zapobiegając ponownej aktywacji komórek przez te same lub podobne patogeny bakteryjne i/lub ich produkty.
IRAK-M
Ekspresja IRAK-M jest zazwyczaj ograniczona do monocytów/makrofagów. Jest indukowana w okresie dojrzewania makrofagów i podczas sygnalizacji TLR/IL-1R [26,46]. Jednak badania przeprowadzone przez Balaciego i wsp. [3] wykazały ekspresję białka IRAK-M w komórkach nabłonka płuc u chorych na astmę. Wyniki te zostały potwierdzone na modelu mysim [50,90].
IRAK-M należy do rodziny IRAK (interleukin-1 receptor-associated kinase), która u ssaków liczy czterech przedstawicieli: IRAK-1, IRAK-2, IRAK-3 (IRAK-M), IRAK-4. Pomimo posiadania 12 serynowo-treoninowych subdomen, IRAK-M nie wykazuje aktywności katalitycznej obserwowanej w IRAK-1, IRAK-4 [103]. Na N-końcu cząsteczki IRAK-M, podobnie jak w pozostałych cząsteczkach IRAK, znajduje się domena DD (death domain) niezbędna do interakcji TIR-TIR z MyD88. IRAK-M może tworzyć heterodimery z IRAK-1 lub IRAK-2 i wiązać zarówno MyD88 jak i TRAF6 [26]. Uważa się, że po przyłączeniu liganda do TLR/IL-1 i utworzeniu kompleksu adaptorowego MyD88/IRAK-4, IRAK-M wiąże się do tego kompleksu, hamując fosforylację IRAK- 1 przez kinazę IRAK-4. Wskutek tego nie dochodzi do utworzenia kolejnego kompleksu TRAF6/IRAK-1, który odpowiada na ścieżce sygnalizacyjnej TLR, za inicjację kinaz IκB(IKK) i MAP, niezbędnych do aktywacji czynników transkrypcyjnych, tj. NF-κB i AP1 [37,46]. Hamuje to wytwarzanie czynników prozapalnych i indukcją tolerancji endotoksycznej.
Ekspresja IRAK-M może być zwiększana lub obniżana w odpowiedzi na liczne cząsteczki sygnalizacyjne zarówno powierzchniowe jak i wewnątrzkomórkowe oraz rozpuszczalne czynniki endogenne i egzogenne [28,29,34,50,123]. Jest to szczególnie istotne w aspekcie klinicznym. Z jednej strony, w przewlekłych zapaleniach ekspresja kinazy IRAK-M jest wręcz pożądana, ponieważ może ograniczać nadmierną odpowiedź immunologiczną, z drugiej zaś IRAK-M może zapobiegać właściwej, wrodzonej odporności.
W przypadku nowotworów, immunosupresyjne mikrośrodowisko guza może powodować indukcję IRAK-M w makrofagach w obrębie guza. U chorych na przewlekłą białaczkę szpikową (chronic myeloid leukemia), w monocytach krwi obwodowej odnotowano podwyższoną ekspresję IRAK-M [16].
Obecnie ocenia się, że białko IRAK-M wykazuje ekspresję w wielu różnych typach komórek odpornościowych i nabłonkowych i może być indukowane przez różne bodźce. To sugeruje, że IRAK-M może odgrywać rolę w regulacji o wiele więcej stanów zapalnych niż dotychczas sądzono.
Podsumowanie
Sygnalizacja z receptorów PRR ma podstawowe znaczenie w inicjowaniu wrodzonej i adaptacyjnej odpowiedzi immunologicznej. Niedostateczna bądź opóźniona reakcja w odpowiedzi na powstały kompleks receptor-ligand, może prowadzić do zwiększonej podatności na infekcje i brak kontroli nad nią, podczas gdy nadmierna aktywacja receptorów może spowodować autoimmunizację lub przewlekłe zapalenie. Sygnalizacja z TLR musi być ściśle regulowana w celu utrzymania równowagi między aktywacją i hamowaniem oraz równoważeniem korzystnych i niekorzystnych skutków rozpoznania antygenu.
Wyniki dotychczasowych badań pozwalają stwierdzić, że sygnały transmitowane z TLR są kontrolowane przez swoiste inhibitory, które w wielu przypadkach wykazują swoistość tkankową i komórkową. Jednak mechanizmy pozwalające zrozumieć tę specyfikę są słabo poznane. Wiele hamujących, znanych już czynników, wpływających na sygnalizację TLR musi jeszcze zostać poddane ocenie, aby obraz homeostazy organizmu na tym odcinku, był pełniejszy. Zrozumienie funkcji kontrolnej negatywnych regulatorów sygnalizacji TLR może być pomocne w oferowaniu nowych możliwości przesunięcia równowagi między tolerancją a odpornością. Wiedza ta mogłaby być wykorzystana do opracowania nowych strategii terapeutycznych wielu chorób, w tym chorób nowotworowych.
PIŚMIENNICTWO
[1] Albiger B., Dahlberg S., Henriques-Normark B., Normark S.: Role of the innate immune system in host defence against bacterial infections: focus on the Toll-like receptors. J. Intern. Med., 2007; 261: 511-528
[PubMed] [Full Text HTML] [Full Text PDF]
[2] Ashkenazi A., Dixit V.M.: Apoptosis control by death and decoy receptors. Curr. Opin. Cell Biol., 1999; 11: 255-260
[PubMed] [Full Text HTML] [Full Text PDF]
[3] Balaci L., Spada M.C., Olla N., Sole G., Loddo L., Anedda F., Naitza S., Zuncheddu M.A., Maschio A., Altea D., Uda M., Pilia S., Sanna S., Masala M., Crisponi L., Fattori M., Devoto M., Doratiotto S., Rassu S., Mereu S., Giua E., Cadeddu N.G., Atzeni R., Pelosi U., Corrias A., Perra R., Torrazza P.L., Pirina P., Ginesu F., Marcias S., Schintu M.G., Del Giacco G.S., Manconi P.E., Malerba G., Bisognin A., Trabetti E., Boner A., Pescollderungg L., Pignatti P.F., Schlessinger D., Cao A., Pilia G.: IRAK-M is involved in the pathogenesis of early-onset persistent asthma. Am. J. Hum. Genet., 2007; 80: 1103-1114
[PubMed] [Full Text HTML] [Full Text PDF]
[4] Balakirev M.Y., Tcherniuk S.O., Jaquinod M., Chroboczek J.: Otubains: a new family of cysteine proteases in the ubiquitin pathway. EMBO Rep., 2003; 4: 517-522
[PubMed] [Full Text HTML] [Full Text PDF]
[5] Beutler B.: Inferences, questions and possibilities in Toll-like receptor signalling. Nature, 2004; 430: 257-263
[PubMed]
[6] Boone D.L., Turer E.E., Lee E.G., Ahmad R.C., Wheeler M.T., Tsui C., Hurley P., Chien M., Chai S., Hitotsumatsu O., McNally E., Pickart C., Ma A.: The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol., 2004; 5: 1052-1060
[PubMed]
[7] Borm M.E., van Bodegraven A.A., Mulder C.J., Kraal G., Bouma G.: The effect of NOD2 activation on TLR2-mediated cytokine responses is dependent on activation dose and NOD2 genotype. Genes Immun., 2008; 9: 274-278
[PubMed] [Full Text HTML] [Full Text PDF]
[8] Brint E.K., Xu D., Liu H., Dunne A., McKenzie A.N., O’Neill L.A., Liew F.Y.: ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat. Immunol., 2004; 5: 373-379
[PubMed]
[9] Brissoni B., Agostini L., Kropf M., Martinon F., Swoboda V., Lippens S., Everett H., Aebi N., Janssens S., Meylan E., Felberbaum-Corti M., Hirling H., Gruenberg J., Tschopp J., Burns K.: Intracellular trafficking of interleukin-1 receptor I requires Tollip. Curr. Biol., 2006; 16: 2265-2270
[PubMed] [Full Text HTML] [Full Text PDF]
[10] Bulut Y., Faure E., Thomas L., Equils O., Arditi M.: Cooperation of Toll-like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1 receptor signaling molecules in Toll-like receptor 2 signaling. J. Immunol., 2001; 167: 987-994
[PubMed] [Full Text HTML] [Full Text PDF]
[11] Burns K., Clatworthy J., Martin L., Martinon F., Plumpton C., Maschera B., Lewis A., Ray K., Tschopp J., Volpe F.: Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat. Cell Biol., 2000; 2: 346-351
[PubMed]
[12] Burns K., Janssens S., Brissoni B., Olivos N., Beyaert R., Tschopp J.: Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J. Exp. Med., 2003; 197: 263-268
[PubMed] [Full Text HTML] [Full Text PDF]
[13] Carty M., Goodbody R., Schröder M., Stack J., Moynagh P.N., Bowie A.G.: The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol., 2006; 7: 1074-1081
[PubMed]
[14] Chamaillard M., Hashimoto M., Horie Y., Masumoto J., Qiu S., Saab L., Ogura Y., Kawasaki A., Fukase K., Kusumoto S., Valvano M.A., Foster S.J., Mak T.W., Nunez G., Inohara N.: An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol., 2003; 4: 702-707
[PubMed]
[15] Couillault C., Pujol N., Reboul J., Sabatier L., Guichou J.F., Kohara Y., Ewbank J.J.: TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat. Immunol., 2004; 5: 488-494
[PubMed]
[16] del Fresno C., Otero K., Gómez-García L., González-León M.C., Soler-Ranger L., Fuentes-Prior P., Escoll P., Baos R., Caveda L., García F., Arnalich F., López-Collazo E.: Tumor cells deactivate human monocytes by up-regulating IL-1 receptor associated kinase-M expression via CD44 and TLR4. J. Immunol., 2005; 174: 3032-3040
[PubMed] [Full Text HTML] [Full Text PDF]
[17] Diehl G.E., Yue H.H., Hsieh K., Kuang A.A., Ho M., Morici L.A., Lenz L.L., Cado D., Riley L.W., Winoto A.: TRAIL-R as a negative regulator of innate immune cell responses. Immunity, 2004; 21: 877-889
[PubMed] [Full Text HTML] [Full Text PDF]
[18] Dimitriou I.D., Clemenza L., Scotter A.J., Chen G., Guerra F.M., Rottapel R.: Putting out the fire: coordinated suppression of the innate and adaptive immune systems by SOCS1 and SOCS3 proteins. Immunol. Rev., 2008; 224: 265-283
[PubMed]
[19] Divanovic S., Trompette A., Atabani S.F., Madan R., Golenbock D.T., Visintin A., Finberg R.W., Tarakhovsky A., Vogel S.N., Belkaid Y., Kurt-Jones E.A., Karp C.L.: Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat. Immunol., 2005; 6: 571-578
[PubMed]
[20] Divanovic S., Trompette A., Petiniot L.K., Allen J.L., Flick L.M., Belkaid Y., Madan R., Haky J.J., Karp C.L.: Regulation of TLR4 signaling and the host interface with pathogens and danger: the role of RP105. J. Leukoc. Biol., 2007; 82: 265-271
[PubMed] [Full Text HTML] [Full Text PDF]
[21] Dixit V.M., Green S., Sarma V., Holzman L.B., Wolf F.W., O’Rourke K., Ward P.A., Prochownik E.V., Marks R.M.: Tumor necrosis factor-α induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J. Biol. Chem., 1990; 265: 2973-2978
[PubMed] [Full Text PDF]
[22] Dunne A., Ejdeback M., Ludidi P.L., O’Neill L.A., Gay N.J.: Structural complementarity of Toll/interleukin-1 receptor domains in Toll-like receptors and the adaptors Mal and MyD88. J. Biol. Chem., 2003; 278: 41443-41451
[PubMed] [Full Text HTML] [Full Text PDF]
[23] Fitzgerald K.A., Chen Z.J.: Sorting out Toll signals. Cell, 2006; 125: 834-836
[PubMed] [Full Text HTML] [Full Text PDF]
[24] Friedman C.S., O’Donnell M.A., Legarda-Addison D., Ng A., Cárdenas W.B., Yount J.S., Moran T.M., Basler C.F., Komuro A., Horvath C.M., Xavier R., Ting A.T.: The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep., 2008; 9: 930-936
[PubMed] [Full Text HTML] [Full Text PDF]
[25] Frobose H., Ronn S.G., Heding P.E., Mendoza H., Cohen P., Mandrup-Poulsen T., Billestrup N.: Suppressor of cytokine Signaling-3 inhibits interleukin-1 signaling by targeting the TRAF-6/TAK1 complex. Mol. Endocrinol., 2006; 20: 1587-1596
[PubMed] [Full Text HTML] [Full Text PDF]
[26] Fukao T., Koyasu S.: PI3K and negative regulation of TLR signaling. Trends Immunol., 2003; 24: 358-363
[PubMed]
[27] Gainetdinov R.R., Premont R.T., Bohn L.M., Lefkowitz R.J., Caron M.G.: Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci., 2004; 27: 107-144
[PubMed] [Full Text HTML] [Full Text PDF]
[28] Hassan F., Islam S., Tumurkhuu G., Dagvadorj J., Naiki Y., Komatsu T., Koide N., Yoshida T., Yokochi T.: Involvement of interleukin-1 receptor-associated kinase (IRAK)-M in toll-like receptor (TLR) 7-mediated tolerance in RAW 264.7 macrophage-like cells. Cell. Immunol., 2009; 256: 99-103
[PubMed]
[29] Hayashi T., Gray C.S., Chan M., Tawatao R.I., Ronacher L., McGargill M.A., Datta S.K., Carson D.A., Corr M.: Prevention of autoimmune disease by induction of tolerance to Toll-like receptor 7. Proc. Natl. Acad. Sci. USA, 2009; 106: 2764-2769
[PubMed] [Full Text HTML] [Full Text PDF]
[30] Heyninck K., De Valck D., Vanden Berghe W., Van Criekinge W., Contreras R., Fiers W., Haegeman G., Beyaert R.: The zinc finger protein A20 inhibits TNF-induced NF-κB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-κB-inhibiting protein ABIN. J. Cell Biol., 1999; 145: 1471-1482
[PubMed] [Full Text HTML] [Full Text PDF]
[31] Hilton D.J.: Negative regulators of cytokine signal transduction. Cell. Mol. Life Sci., 1999; 55: 1568-1577
[PubMed]
[32] Hilton D.J., Richardson R.T., Alexander W.S., Viney E.M., Willson T.A., Sprigg N.S., Starr R., Nicholson S.E., Metcalf D., Nicola N.A.: Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA, 1998; 95: 114-119
[PubMed] [Full Text PDF]
[33] Horng T., Barton G.M., Flavell R.A., Medzhitov R.: The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature, 2002; 420: 329-333
[PubMed]
[34] Hubbard L.L., Ballinger M.N., Thomas P.E., Wilke C.A., Standiford T.J., Kobayashi K.S., Flavell R.A., Moore B.B.: A role for IL-1 receptor-associated kinase-M in prostaglandin E2-induced immunosuppression post-bone marrow transplantation. J. Immunol., 2010; 184: 6299-6308
[PubMed] [Full Text HTML] [Full Text PDF]
[35] Ilangumaran S., Ramanathan S., Rottapel R.: Regulation of the immune system by SOCS family adaptor proteins. Semin. Immunol., 2004; 16: 351-365
[PubMed]
[36] Iwami K.I., Matsuguchi T., Masuda A., Kikuchi T., Musikacharoen T., Yoshikai Y.: Cutting edge: naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J. Immunol., 2000; 165: 6682-6686
[PubMed] [Full Text HTML] [Full Text PDF]
[37] Janssens S., Beyaert R.: Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol. Cell, 2003; 11: 293-302
[PubMed] [Full Text HTML] [Full Text PDF]
[38] Janssens S., Burns K., Tschopp J., Beyaert R.: Regulation of interleukin-1- and lipopolysaccharide-induced NF-κB activation by alternative splicing of MyD88. Curr. Biol., 2002; 12: 467-471
[PubMed] [Full Text HTML] [Full Text PDF]
[39] Janssens S., Burns K., Vercammen E., Tschopp J., Beyaert R.: MyD88S, a splice variant of MyD88, differentially modulates NF-κB- and AP-1-dependent gene expression. FEBS Lett., 2003; 548: 103-107
[PubMed] [Full Text HTML] [Full Text PDF]
[40] Jeong E., Lee J.Y.: Intrinsic and extrinsic regulation of innate immune receptors. Yonsei Med. J., 2011; 52: 379-392
[PubMed] [Full Text PDF]
[41] Jiang Z., Zamanian-Daryoush M., Nie H., Silva A.M., Williams B.R., Li X.: Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFκB and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J. Biol. Chem., 2003; 278, 16713-16719
[PubMed] [Full Text HTML] [Full Text PDF]
[42] Kagan J.C., Medzhitov R.: Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell, 2006; 125: 943-955
[PubMed]
[43] Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K.J., Takeuchi O., Akira S.: IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol., 2005; 6: 981-988
[PubMed]
[44] Kile B.T., Schulman B.A., Alexander W.S., Nicola N.A., Martin H.M., Hilton D.J.: The SOCS box: a tale of destruction and degradation. Trends Biochem. Sci., 2002; 27: 235-241
[PubMed]
[45] Kinjyo I., Hanada T., Inagaki-Ohara K., Mori H., Aki D., Ohishi M., Yoshida H., Kubo M., Yoshimura A.: SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity, 2002; 17: 583-591
[PubMed]
[46] Kobayashi K., Hernandez L.D., Galán J.E., Janeway C.A. Jr., Medzhitov R., Flavell R.A.: IRAK-M is a negative regulator of Toll-like receptor signaling. Cell, 2002; 110: 191-202
[PubMed]
[47] Kubo M., Hanada T., Yoshimura A.: Suppressors of cytokine signaling and immunity Nat. Immunol., 2003; 4: 1169-1176
[PubMed]
[48] Kulczycka L., Sysa-Jędrzejowska A., Robak E.: Udział receptorów Toll-like w patogenezie wybranych chorób skóry. Postępy Hig. Med. Dośw., 2010; 64: 364-371
[PubMed] [Full Text HTML] [Full Text PDF]
[49] Kumar H., Kawai T., Akira S.: Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun., 2009; 388: 621-625
[PubMed]
[50] Lagler H., Sharif O., Haslinger I., Matt U., Stich K., Furtner T., Doninger B., Schmid K., Gattringer R., de Vos A.F., Knapp S.: TREM-1 activation alters the dynamics of pulmonary IRAK-M expression in vivo and improves host defense during pneumococcal pneumonia. J. Immunol., 2009; 183: 2027-2036
[PubMed] [Full Text HTML] [Full Text PDF]
[51] LeBouder E., Rey-Nores J.E., Rushmere N.K., Grigorov M., Lawn S.D., Affolter M., Griffin G.E., Ferrara P., Schiffrin E.J., Morgan B.P., Labéta M.O.: Soluble forms of Toll-like receptor (TLR)2 capable of modulating TLR2 signaling are present in human plasma and breast milk. J. Immunol., 2003; 171: 6680-6689
[PubMed] [Full Text HTML] [Full Text PDF]
[52] Lécart S., Lecointe N., Subramaniam A., Alkan S., Ni D., Chen R., Boulay V., Pene J., Kuroiwa K., Tominaga S., Yssel H.: Activated, but not resting human Th2 cells, in contrast to Th1 and T regulatory cells, produce soluble ST2 and express low levels of ST2L at the cell surface. Eur. J. Immunol., 2002; 32: 2979-2987
[PubMed]
[53] Lefkowitz R.J., Whalen E.J.: β-arrestins: traffic cops of cell signaling. Curr. Opin. Cell Biol., 2004; 16: 162-168
[PubMed]
[54] Li L., Soetandyo N., Wang Q., Ye Y.: The zinc finger protein A20 targets TRAF2 to the lysosomes for degradation. Biochim. Biophys. Acta, 2009; 1793: 346-353
[PubMed] [Full Text HTML] [Full Text PDF]
[55] Li T., Hu J., Li L.: Characterization of Tollip protein upon lipopolysaccharide challenge. Mol. Immunol., 2004; 41: 85-92
[PubMed]
[56] Liew F.Y., Xu D., Brint E.K., O’Neill L.A.: Negative regulation of Toll-like receptor-mediated immune responses. Nat. Rev. Immunol., 2005; 5: 446-458
[PubMed]
[57] Lin S.C., Chung J.Y., Lamothe B., Rajashankar K., Lu M., Lo Y.C., Lam A.Y., Darnay B.G., Wu H.: Molecular basis for the unique deubiquitinating activity of the NF-κB inhibitor A20. J. Mol. Biol., 2008; 376: 526-540
[PubMed] [Full Text HTML] [Full Text PDF]
[58] Liu J., Buckley J.M., Redmond H.P., Wang J.H.: ST2 negatively regulates TLR2 signaling, but is not required for bacterial lipoprotein-induced tolerance. J. Immunol., 2010; 184: 5802-5808
[PubMed] [Full Text HTML] [Full Text PDF]
[59] Lo Y.L., Beckhouse A.G., Boulus S.L., Wells C.A.: Diversification of TOLLIP isoforms in mouse and man. Mamm. Genome, 2009; 20: 305-314
[PubMed]
[60] Luttrell L.M., Roudabush F.L., Choy E.W., Miller W.E., Field M.E.,Pierce K.L., Lefkowitz R.J.: Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. USA, 2001; 98: 2449-2454
[PubMed] [Full Text HTML] [Full Text PDF]
[61] Mabilleau G., Chappard D., Sabokbar A.: Role of the A20-TRAF6 axis in lipopolysaccharide-mediated osteoclastogenesis. J. Biol. Chem., 2011; 286: 3242-3249
[PubMed] [Full Text HTML] [Full Text PDF]
[62] Mansell A., Smith R., Doyle S.L., Gray P., Fenner J.E., Crack P.J., Nicholson S.E., Hilton D.J., O’Neill L.A., Hertzog P.J.: Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat. Immunol., 2006; 7: 148-155
[PubMed]
[63] Martin T.F.: Phosphoinositide lipids as signaling molecules: common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu. Rev. Cell Dev. Biol., 1998; 14: 231-264
[PubMed]
[64] Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J.: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature, 2006; 440: 237-241
[PubMed] [Full Text HTML] [Full Text PDF]
[65] McDonald P.H., Cote N.L., Lin F.T., Premont R.T., Pitcher J.A., Lefkowitz R.J.: Identification of NSF as a β-arrestin1-binding protein. Implications for β2-adrenergic receptor regulation. J. Biol. Chem., 1999; 274: 10677-10680
[PubMed] [Full Text HTML] [Full Text PDF]
[66] McGettrick, A.F., O’Neill L.A.: The expanding family of MyD88-like adaptors in Toll-like receptor signal transduction. Mol. Immunol., 2004; 41: 577-582
[PubMed]
[67] Medzhitov R.: Recognition of microorganisms and activation of the immune response. Nature, 2007; 449: 819-826
[PubMed]
[68] Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J.: Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature, 2005; 437: 1167-1172
[PubMed]
[69] Meylan E., Tschopp J.: Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol. Cell, 2006; 22: 561-569
[PubMed] [Full Text HTML] [Full Text PDF]
[70] Meylan E., Tschopp J., Karin M.: Intracellular pattern recognition receptors in the host response. Nature, 2006; 442: 39-44
[PubMed]
[71] Mink M., Fogelgren B., Olszewski K., Maroy P., Csiszar K.: A novel human gene (SARM) at chromosome 17q11 encodes a protein with a SAM motif and structural similarity to Armadillo/β-catenin that is conserved in mouse, Drosophila, and Caenorhabditis elegans. Genomics, 2001; 74: 234-244
[PubMed]
[72] Miyake K., Yamashita Y., Ogata M., Sudo T., Kimoto M.: RP105, a novel B cell surface molecule implicated in B cell activation, is a member of the leucine-rich repeat protein family. J. Immunol., 1995; 154: 3333-3340
[PubMed]
[73] Nagai Y., Kobayashi T., Motoi Y., Ishiguro K., Akashi S., Saitoh S., Kusumoto Y., Kaisho T., Akira S., Matsumoto M., Takatsu K., Miyake K.: The radioprotective 105/MD-1 complex links TLR2 and TLR4/MD-2 in antibody response to microbial membranes. J. Immunol., 2005; 174: 7043-7049
[PubMed] [Full Text HTML] [Full Text PDF]
[74] Nakagawa R., Naka T., Tsutsui H., Fujimoto M., Kimura A., Abe T., Seki E., Sato S., Takeuchi O., Takeda K., Akira S., Yamanishi K., Kawase I., Nakanishi K., Kishimoto T.: SOCS-1 participates in negative regulation of LPS responses. Immunity, 2002; 17: 677-687
[PubMed]
[75] Nallagatla S.R., Hwang J., Toroney R., Zheng X., Cameron C.E., Bevilacqua P.C.: 5′-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science, 2007; 318: 1455-1458
[PubMed]
[76] O’Neill L.A., Fitzgerald K.A., Bowie A.G.: The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol., 2003; 24: 286-290
[PubMed]
[77] Oguro A., Kubota H., Shimizu M., Ishiura S., Atomi Y.: Protective role of the ubiquitin binding protein Tollip against the toxicity of polyglutamine-expansion proteins. Neurosci. Lett., 2011; 503: 234-239
[PubMed]
[78] Opipari A.W. Jr., Boguski M.S., Dixit V.M.: The A20 cDNA induced by tumor necrosis factor α encodes a novel type of zinc finger protein. J. Biol. Chem., 1990; 265: 14705-14708
[PubMed] [Full Text PDF]
[79] Park J.H., Kim Y.G., McDonald C., Kanneganti T.D., Hasegawa M., Body-Malapel M., Inohara N., Núnez G.: RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J. Immunol., 2007; 178: 2380-2386
[PubMed] [Full Text HTML] [Full Text PDF]
[80] Pauleau A.L., Murray P.J.: Role of nod2 in the response of macrophages to toll-like receptor agonists. Mol. Cell. Biol., 2003; 23: 7531-7539
[PubMed] [Full Text HTML] [Full Text PDF]
[81] Peng J., Yuan Q., Lin B., Panneerselvam P., Wang X., Luan X.L., Lim S.K., Leung B.P., Ho B., Ding J.L.: SARM inhibits both TRIF- and MyD88-mediated AP-1 activation. Eur. J. Immunol., 2010; 40: 1738-1747
[PubMed] [Full Text HTML] [Full Text PDF]
[82] Perry S.J., Baillie G.S., Kohout T.A., McPhee I., Magiera M.M., Ang K.L., Miller W.E., McLean A.J., Conti M., Houslay M.D., Lefkowitz R.J.: Targeting of cyclic AMP degradation to β2-adrenergic receptors by β-arrestins. Science, 2002; 298: 834-836
[PubMed]
[83] Pitti R.M., Marsters S.A., Ruppert S., Donahue C.J., Moore A., Ashkenazi A.: Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem., 1996; 271: 12687-12690
[PubMed] [Full Text HTML] [Full Text PDF]
[84] Qin J., Qian Y., Yao J., Grace C., Li X.: SIGIRR inhibits interleukin-1 receptor- and Toll-like receptor 4-mediated signaling through different mechanisms. J. Biol. Chem., 2005; 280: 25233-25241
[PubMed] [Full Text HTML] [Full Text PDF]
[85] Rothenfusser S., Goutagny N., DiPerna G., Gong M., Monks B.G., Schoenemeyer A., Yamamoto M., Akira S., Fitzgerald K.A.: The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol., 2005; 175: 5260-5268
[PubMed] [Full Text HTML] [Full Text PDF]
[86] Ryo A., Suizu F., Yoshida Y., Perrem K., Liou Y.C., Wulf G., Rottapel R., Yamaoka S., Lu K.P.: Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell, 2003; 12: 1413-1426
[PubMed]
[87] Sasaki A., Yasukawa H., Shouda T., Kitamura T., Dikic I., Yoshimura A.: CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J. Biol. Chem., 2000; 275: 29338-29347
[PubMed] [Full Text HTML] [Full Text PDF]
[88] Sato S., Sugiyama M., Yamamoto M., Watanabe Y., Kawai T., Takeda K., Akira S.: Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κB and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol., 2003; 171, 4304-4310
[PubMed] [Full Text HTML] [Full Text PDF]
[89] Schaefer U., Voloshanenko O., Willen D., Walczak H.: TRAIL: a multifunctional cytokine. Front. Biosci., 2007; 12: 3813-3824
[PubMed]
[90] Seki M., Kohno S., Newstead M.W., Zeng X., Bhan U., Lukacs N.W., Kunkel S.L., Standiford T.J.: Critical role of IL-1 receptor-associated kinase-M in regulating chemokine-dependent deleterious inflammation in murine influenza pneumonia. J. Immunol., 2010; 184: 1410-1418
[PubMed] [Full Text HTML] [Full Text PDF]
[91] Seth R.B., Sun L., Chen Z.J.: Antiviral innate immunity pathways. Cell Res., 2006; 16: 141-147
[PubMed] [Full Text HTML] [Full Text PDF]
[92] Seth R.B., Sun L., Ea C.K., Chen Z.J.: Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell, 2005; 122: 669-682
[PubMed]
[93] Sims J.E., Smith D.E.: The IL-1 family: regulators of immunity. Nat. Rev. Immunol., 2010; 10: 89-102
[PubMed]
[94] So E.Y., Ouchi T.: The application of Toll like receptors for cancer therapy. Int. J. Biol. Sci., 2010; 6: 675-681
[PubMed] [Full Text HTML] [Full Text PDF]
[95] Sweet M.J., Leung B.P., Kang D., Sogaard M., Schulz K., Trajkovic V., Campbell C.C., Xu D., Liew F.Y.: A novel pathway regulating lipopolysaccharide-induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expression. J. Immunol., 2001; 166: 6633-6639
[PubMed] [Full Text HTML] [Full Text PDF]
[96] Takahasi K., Yoneyama M., Nishihori T., Hirai R., Kumeta H., Narita R., Gale M. Jr., Inagaki F., Fujita T.: Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell, 2008; 29: 428-440
[PubMed]
[97] Takeuchi O., Akira S.: MDA5/RIG-1 and virus recognition. Curr. Opin. Immunol., 2008; 20: 17-22
[PubMed] [Full Text HTML] [Full Text PDF]
[98] Takezako N., Hayakawa M., Hayakawa H., Aoki S., Yanagisawa K., Endo H., Tominaga S.: ST2 suppresses IL-6 production via the inhibition of IκB degradation induced by the LPS signal in THP-1 cells. Biochem. Biophys. Res. Commun., 2006; 341: 425-432
[PubMed]
[99] Turer E.E., Tavares R.M., Mortier E., Hitotsumatsu O., Advincula R., Lee B., Shifrin N., Malynn B.A., Ma A.: Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J. Exp. Med., 2008; 205: 451-464
[PubMed] [Full Text HTML] [Full Text PDF]
[100] Vereecke L., Sze M., Mc Guire C., Rogiers B., Chu Y., Schmidt-Supprian M., Pasparakis M., Beyaert R., van Loo G.: Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J. Exp. Med., 2010; 207: 1513-1523
[PubMed] [Full Text HTML] [Full Text PDF]
[101] Verstrepen L., Verhelst K., van Loo G., Carpentier I., Ley S.C., Beyaert R.: Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem. Pharmacol., 2010; 80: 2009-2020
[PubMed]
[102] Walczak H., Miller R.E., Ariail K., Gliniak B., Griffith T.S., Kubin M., Chin W., Jones J., Woodward A., Le T., Smith C., Smolak P., Goodwin R.G., Rauch C.T., Schuh J.C., Lynch D.H.: Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med., 1999; 5: 157-163
[PubMed]
[103] Wang J., Hu Y., Deng W.W., Sun B.: Negative regulation of Toll-like receptor signaling pathway. Microbes Infect., 2009; 11: 321-327
[PubMed]
[104] Wang Y., Tang Y., Teng L., Wu Y., Zhao X., Pei G.: Association of β-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol., 2006; 7: 139-147
[PubMed]
[105] Watanabe T., Kitani A., Murray P.J., Strober W.: NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat. Immunol., 2004; 5: 800-808
[PubMed]
[106] Weber F., Wagner V., Rasmussen S.B., Hartmann R., Paludan S.R.: Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol., 2006; 80: 5059-5064
[PubMed] [Full Text HTML] [Full Text PDF]
[107] Wertz I.E., O’Rourke K.M., Zhou H., Eby M., Aravind L., Seshagiri S., Wu P., Wiesmann C., Baker R., Boone D.L., Ma A., Koonin E.V., Dixit V.M.: De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature, 2004; 430: 694-699
[PubMed]
[108] Wiley S.R., Schooley K., Smolak P.J., Din W.S., Huang C.P., Nicholl J.K., Sutherland G.R., Smith T.D., Rauch C., Smith C.A., Rauch Ch., Smith C.A., Goodwin R.G.: Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity, 1995; 3: 673-682
[PubMed]
[109] Windheim M., Lang C., Peggie M., Plater L.A., Cohen P.: Molecular mechanisms involved in the regulation of cytokine production by muramyl dipeptide. Biochem. J., 2007; 404: 179-190
[PubMed] [Full Text HTML] [Full Text PDF]
[110] Witherow D.S., Garrison T.R., Miller W.E., Lefkowitz R.J.: β-arrestin inhibits NF-κB activity by means of its interaction with the NF-κB inhibitor IκBα. Proc. Natl. Acad. Sci. USA, 2004; 101: 8603-8607
[PubMed] [Full Text HTML] [Full Text PDF]
[111] Xiao H., Gulen M.F., Qin J., Yao J., Bulek K., Kish D., Altuntas C.Z., Wald D., Ma C., Zhou H., Tuohy V.K., Fairchild R.L., de la Motte C., Cua D., Vallance B.A., Li X.: The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity, 2007; 26: 461-475
[PubMed]
[112] Xu L.G., Wang Y.Y., Han K.J., Li L.Y., Zhai Z., Shu H.B.: VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell, 2005; 19: 727-740
[PubMed] [Full Text HTML] [Full Text PDF]
[113] Yamakami M., Yoshimori T., Yokosawa H.: Tom1, a VHS domain-containing protein, interacts with tollip, ubiquitin, and clathrin. J. Biol. Chem., 2003; 278: 52865-52872
[PubMed] [Full Text HTML] [Full Text PDF]
[114] Yamamoto M., Sato S., Hemmi H., Sanjo H., Uematsu S., Kaisho T., Hoshino K., Takeuchi O., Kobayashi M., Fujita T., Takeda K., Akira S.: Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature, 2002; 420: 324-329
[PubMed]
[115] Yasukawa H., Misawa H., Sakamoto H., Masuhara M., Sasaki A., Wakioka T., Ohtsuka S., Imaizumi T., Matsuda T., Ihle J.N., Yoshimura A.: The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J., 1999; 18: 1309-1320
[PubMed] [Full Text HTML] [Full Text PDF]
[116] Yin H., Huang B.J., Yang H., Huang Y.F., Xiong P., Zheng F., Chen X.P., Chen Y.F., Gong F.L.: Pretreatment with soluble ST2 reduces warm hepatic ischemia/reperfusion injury. Biochem. Biophys. Res. Commun., 2006; 351: 940-946
[PubMed]
[117] Yoneyama M., Fujita T.: Function of RIG-I-like receptors in antiviral innate immunity. J. Biol. Chem., 2007; 282: 15315-15318
[PubMed] [Full Text HTML] [Full Text PDF]
[118] Yoneyama M., Kikuchi M., Matsumoto K., Imaizumi T., Miyagishi M., Taira K., Foy E., Loo Y.M., Gale M. Jr., Akira S., Yonehara S., Kato A., Fujita T.: Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol., 2005; 175: 2851-2858
[PubMed] [Full Text HTML] [Full Text PDF]
[119] Yoshida H., Jono H., Kai H., Li J.D.: The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for Toll-like receptor 2 signaling via negative cross-talk with TRAF6 and TRAF7 J. Biol. Chem., 2005; 280, 41111-41121
[PubMed] [Full Text HTML] [Full Text PDF]
[120] Yoshimura A., Naka T., Kubo M.: SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol., 2007; 7: 454-465
[PubMed]
[121] Yuk J.M., Jo E.K.: Toll-like receptors and innate immunity. J. Bacteriol. Virol., 2011; 41: 225- 235
[Abstract] [Full Text PDF]
[122] Yuk J.M., Shin D.M., Lee H.M., Kim J.J., Kim S.W., Jin H.S., Yang C.S., Park K.A., Chanda D., Kim D.K., Huang S.M., Lee S.K., Lee C.H., Kim J.M., Song C.H., Lee S.Y., Hur G.M., Moore D.D., Choi H.S., Jo.EK.: The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol., 2011; 12: 742-751
[PubMed]
[123] Zacharioudaki V., Androulidaki A., Arranz A., Vrentzos G., Margioris A.N., Tsatsanis C.: Adiponectin promotes endotoxin tolerance in macrophages by inducing IRAK-M expression. J. Immunol., 2009; 182: 6444-6451
[PubMed] [Full Text HTML] [Full Text PDF]
[124] Zhang G., Ghosh S.: Negative regulation of toll-like receptor-mediated signaling by Tollip. J. Biol. Chem., 2002; 277: 7059-7065
[PubMed] [Full Text HTML] [Full Text PDF]
[125] Zhang S., Guo D., Jiang L., Zhang Q., Qiu X., Wang E.: SOCS3 inhibiting migration of A549 cells correlates with PYK2 signaling in vitro. BMC Cancer, 2008; 8: 150
[PubMed] [Full Text HTML] [Full Text PDF]
Autorki deklarują brak potencjalnych konfliktów interesów.