
Perspektywy wykorzystania interferencji RNA w terapii chorób związanych z zaburzeniami alternatywnego składania RNA
Daniel Wysokiński 1 , Janusz Błasiak 1Streszczenie
Pierwotny transkrypt genu eukariotycznego (pre-mRNA) składa się z regionów kodujących – eksonów, poprzedzielanych sekwencjami niekodującymi intronami, które usuwane są w procesie składania RNA, co prowadzi do powstania dojrzałego transkryptu. Alternatywne składanie pre-mRNA, do niedawna uważane za szczególny wyraz metabolizmu kwasów nukleinowych, odpowiada za dużą różnorodność komórkowego proteomu i służy efektywnemu wykorzystaniu informacji zawartej w genomie. Pełni istotne role w regulacji tak ważnych procesów w organizmie, jak regulacja apoptozy, czy rozwój i plastyczność układu nerwowego. Zadaniem alternatywnego składania jest różnicowe, zależne od warunków i typu komórki składanie mRNA, co w rezultacie daje różniące się strukturą transkrypty tego samego genu, a po translacji różniące się między sobą izoformy białka. Ze względu na złożoność tego mechanizmu alternatywne składanie jest szczególnie podatne na zaburzenia. Niebezpieczne są zwłaszcza zaburzenia wynikające z mutacji w sekwencjach kluczowych dla regulacji alternatywnego składania. Patogeneza wielu schorzeń wynika z zaburzeń w alternatywnym składaniu, a dotyczy to tak różnych stanów chorobowych, jak nowotwory czy choroby neurodegeneracyjne. Z powodu genetycznych korzeni tych schorzeń, ich terapia jest problematyczna. Obecnie duże nadzieje w terapii tych schorzeń wiąże się z zastosowaniem interferencji RNA. Interferencja RNA jest powszechnym u eukariontów mechanizmem wyciszania genów, a zakończone sukcesem próby wyciszania produkcji transkryptów kodujących wadliwe izoformy białek dają nadzieję na opracowanie nowych, skutecznych metod korekcji zaburzeń alternatywnego składania.
Słowa kluczowe: składanie RNA • składanie alternatywne • interferencja RNA
Summary
The primary transcript of an eukaryotic gene (pre-mRNA) is composed of coding regions – exons intervened by non-coding introns – which are removed in the RNA splicing process, leading to the formation of mature, intron-free mRNA. Alternative splicing of pre-mRNA is responsible for high complexity of the cellular proteome and expresses effective use of genetic information contained in genomic DNA. Alternative splicing plays important roles in the organism, including apoptosis regulation or development and plasticity of the nervous system. The main role of alternative splicing is differential, dependent on conditions and the cell type, splicing of mRNA, generating diverse transcripts from one gene, and, after the translation, different isoforms of a particular protein. Because of the high complexity of this mechanism, alternative splicing is particularly prone to errors. The perturbations resulting from mutations in the key sequences for splicing regulations are especially harmful. The pathogenesis of numerous diseases results from disturbed alternative RNA splicing, and those include cancers and neurodegenerative disorders. The treatment of these conditions is problematic due to their genetic background and currently RNA interference, which is a common mechanism of eukaryotic gene regulation, is being studied. Initial successes in the attempts of silencing the expression of faulty protein isoforms support the idea of using RNA interference in targeting disease related to disturbances in alternative splicing of RNA.
Key words:RNA splicing • alternative splicing • RNA interference
Składanie pre-mRNA
Droga od genu do białka jest wieloetapowa. Podczas transkrypcji genu kodującego białko u organizmów eukariotycznych powstaje transkrypt pierwotny – pre-mRNA, który zanim będzie gotowy do transportu poza jądro komórki i weźmie udział w wytwarzaniu białka w procesie translacji, poddany musi zostać wielu modyfikacjom [30] (ryc.1). Pierwotny produkt transkrypcji kodujący białko u organizmów wyższych składa się z eksonów – sekwencji kodujących, poprzedzielanych sekwencjami niekodującymi – intronami. Introny ulegają wycinaniu z pre-mRNA w procesie składania mRNA [100]. Poza usunięciem intronów pre-mRNA poddawany jest też modyfikacjom na obu swoich końcach. Na końcu 5′ takiego transkryptu zachodzi proces „czapeczkowania” (capping) – dołączenie guanozyny wiązaniem 5′,5′-trifosforanowym oraz jej metylacja w pozycji 7. Czapeczka 5′ (7-mG) zabezpiecza transkrypt przed enzymatyczną degradacją przez egzonukleazy 5′-3′, a także jest niezbędna dla transportu mRNA do cytoplazmy i inicjacji procesu translacji [30,100]. Koniec 3′ cząsteczki RNA podlega natomiast cięciu w miejscu poprzedzonym o 10-30 nukleotydów przez sekwencję sygnałową (AAUAAA u ssaków), a następnie ulega poliadenylacji w procesie, w którym polimeraza poliA (PAP) dołącza adenozyny na końcu 3′ cząsteczki, tworząc „ogon poliA” [30,75]. Oba te procesy są konieczne do prawidłowego eksportu mRNA do cytoplazmy oraz efektywnej translacji [103]. W procesie wycinania intronów bierze udział złożona struktura nazywana spliceosomem (spliceosome). Szacuje się, że spliceosom, jako zwarta struktura, zawiera ponad 200 białek i 5 cząsteczek krótkiego (małego) jądrowego RNA (snRNA – small nuclear RNA) [84,91], biorących udział w składaniu mRNA. Jest to zorganizowany, a zarazem dynamiczny zbiór cząsteczek, w którym występują oddziaływania białko-białko, białko-RNA i RNA-RNA [62]. Wśród składników spliceosomu zasadniczą rolę w procesie wycinania intronów pełni pięć małych jądrowych rybonukleoprotein snRNP – U1, U2, U4, U5 i U6, składających się z snRNA oraz stowarzyszonych z nimi białek [78,91]. Proces biogenezy cząsteczek UsnRNP jest również procesem złożonym i przebiega wieloetapowo zarówno w jądrze, jak i cytoplazmie, z udziałem domen jądrowych – ciał Cajala [90,94,113]. Mechanizm wycinania intronów przebiega w kilku etapach, którym towarzyszy wiele rearanżacji w obrębie spliceosomu oraz zmian konformacyjnych czynników biorących udział w tym procesie. Pierwszym etapem składania jest rozpoznanie końca 5′ intronu i miejsca rozgałęzienia (branch point) w intronie, charakteryzującego się zazwyczaj obecnością adeniny, przez czynniki U1 i U2 snRNP. Powstaje w ten sposób kompleks A. Kompleks B powstaje przez dołączenie kompleksu U4•U6/U5 tri-snRNP. Ostatecznie, w wyniku rearanżacji uwalniającej czynniki U1 i U4 snRNP powstaje katalitycznie aktywny kompleks C [91]. W pierwszym etapie wycinania intronu grupa 2′-OH nukleozydu w miejscu rozgałęzienia przeprowadza nukleofilowy atak na wiązanie fosfodiestrowe ostatniego nukleotydu (w eksonie) po stronie 5′ intronu, w wyniku czego powstaje struktura pętli. Następnie grupa 3′-OH wolnego końca eksonu atakuje ostatni nukleotyd intronu po stronie 3′, intron wypętla się tworząc strukturę przypominającą lasso, po czym wolne końce eksonów ulegają połączeniu [83,91,102]. Warto odnotować, iż elementami spliceosomu uczestniczącymi bezpośrednio w katalizie wycinania intronu są cząsteczki RNA. Podobnie jak w szczególnym przypadku autokatalitycznych, samowycinajacych się intronów, tak i w klasycznym składaniu transkryptów RNA odpowiada za katalizę reakcji chemicznej, działając jako rybozym [37,101]. Liczne dane dowodzą ścisłego związku między składaniem pre-mRNA a transkrypcją. Proces składania zachodzi zarówno kotranskrypcyjnie, jak i posttranskrypcyjnie. Ma to szczególne znaczenie w przypadku syntezy i obróbki bardzo długich transkryptów, np. w przypadku genu dystrofiny (2,4 mln par zasad) [39]. Zaznaczyć należy jednak, że składanie kotranskrypcyjne nie jest regułą. Nie wyjaśniono dotąd mechanizmu, który decydowałby o tym, które introny są wycinane kotranskrypcyjnie, a które posttranskrypcyjnie, jednak dane na ten temat pozwalają sądzić, iż nie jest to proces przypadkowy i istnieje tu swoista regulacja [39]. Ponadto, wiele danych wskazuje na ścisły funkcjonalny związek między transkrypcją i składaniem pierwotnego transkryptu. Główną rolę może tu odgrywać C-końcowa domena polimerazy II RNA – domena CTD (C-terminal domain). Brak funkcjonalnej domeny CTD skutkuje silnym zaburzeniem procesu składania RNA i obróbki na obu końcach pierwotnego transkryptu (czapeczkowanie końca 5′, trawienie i poliadenylacja końca 3′) [52,56]. Efekt ten zależny jest w znacznej mierze od stanu fosforylacji domeny CTD. Prawidłowy wzorzec fosforylacji domeny CTD jest niezbędny do prawidłowego przebiegu zarówno wycinania intronów, jak i obróbki obu końców transkryptu. Natura tej regulacji nie została ostatecznie wyjaśniona, jednak wiadomo, że wiele czynników zaangażowanych w proces dojrzewania transkryptu może bezpośrednio oddziaływać z domeną CTD polimerazy II RNA, co prawdopodobnie jest ściśle kontrolowane przez posttranslacyjne modyfikacje domeny CTD [30,71].

Ryc. 1. Uproszczony schemat kanonicznego składania RNA. Fragment pierwotnego transkryptu (pre-mRNA) zawierający intron jest celem dla kompleksu składającego (spliceosomu). Składniki spliceosomu formują aktywny kompleks na pre-mRNA, następuje wycięcie intronu – powstanie struktury „lassa”, po czym składniki spliceosomu oddysocjowują od mRNA. U1, U2, U4, U5, U6 – rybonukleoproteiny, składniki kompleksu składania RNA
Składanie alternatywne i jego regulacja
Przez długi czas uważano, że złożoność fenotypowa organizmu warunkowana jest liczbą genów w genomie tego organizmu. W trakcie poznawania genomu człowieka oraz genomów innych organizmów wątpliwości budził brak bezpośredniej zależności między złożonością organizmu a liczbą genów w jego genomie [41,95]. Intrygującym było zwłaszcza to, iż szacowana liczba genów u człowieka (około 30 000) jest znacznie niższa od szacowanej liczby białek. [79,95]. Doprowadziło to do odkrycia w 1980 r. mechanizmu alternatywnego składania pre-mRNA, kiedy zaobserwowano, że dwa różne białka – jedno związane z błoną komórkową i drugie wydzielane z komórki, są kodowane przez jeden gen [1]. Było to początkiem trwających do dziś badań nad kolejnym poziomem złożoności genomu. Nieprzerwanie identyfikowane są kolejne izoformy znanych białek, a frakcja genów o więcej niż jednym eksonie ulegających alternatywnemu składaniu w ciągu kilku lat wzrosła prawie z 50 do 95-100% [27,64,87].
Alternatywne składanie prowadzi do generowania kilku białek różniących się strukturą (ryc. 2). Jest to proces złożony i jednostkowo dzieli się na kilka zasadniczych typów. Wśród nich wyróżnia się pominięcie/wycięcie eksonu (exon skipping); ekson zostaje wykluczony z mRNA, zachowanie intronu (intron retention) – intron nie zostaje wycięty z pre-mRNA, alternatywne miejsce 3′ składania i 5′ składania – alternatywne miejsca wycięcia intronu, przez co pozostały odcinek – ekson, może mieć różną długość, eksony wzajemnie wykluczające się (mutually exclusive exons) eksony takie nigdy nie występują w dojrzałym transkrypcie razem (zachowany zostaje jeden) [64,79,38]. Zidentyfikowano również alternatywne miejsca startu transkrypcji, alternatywne miejsca poliadenylacji [49], a także alternatywne regiony nieulegające translacji (regiony UTR – untranslated region) [32]. W niektórych przypadkach proces alternatywnego składania może generować eksony o niepodzielnej przez 3 liczbie par zasad (eksony niesymetryczne), co powoduje przesunięcie ramki odczytu i w konsekwencji zmienia właściwości całej dalszej części transkryptu [77]. W jednym genie może zachodzić jeden wariant alternatywnego składania lub też kilka różnych jego typów jednocześnie, dając dużą różnorodność form białka kodowanego przez ten gen [79].

Ryc. 2. Wybrane mechanizmy niekanonicznego składania RNA – składania alternatywnego (dokładny opis w tekście)
Zidentyfikowano wiele genów o bardzo dużym bogactwie białkowych izoform. Przykładem takiego genu jest Dscam u muszki owocowej Drosophila melanogaster. W skład dojrzałego transkryptu tego genu wchodzą 24 eksony. Gen Dscam zawiera jednak 115 eksonów, z czego 95 pogrupowanych w 4 kasety stanowią warianty alternatywne. Tylko jeden ekson z każdej kasety wchodzi w skład końcowego mRNA [72,85]. Taka złożoność genu daje 38 016 potencjalnie możliwych izoform białka – dwa razy więcej niż liczba wszystkich genów. Produkty genu Dscam pełnią istotne role w ośrodkowym układzie nerwowym, m.in. w morfogenezie komórek nerwowych i tworzeniu synaps w mózgu [20,25]. Na obecnym poziomie wiedzy wydaje się, że układ nerwowy charakteryzuje się szczególnie wysokim poziomem alternatywnego składania, co skłaniać może do wniosku, iż po części to ten mechanizm jest odpowiedzialny za wyjątkową złożoność i zarazem plastyczność i dynamiczną naturę mózgu [109].
Ludzki gen KCNMA1, kodujący podjednostkę kanału potasowego zależnego od wapnia, jest kolejnym przykładem genu generującego dużą liczbę izoform białkowych. Transkrypt tego genu może podlegać alternatywnemu składaniu na kilka sposobów obejmujących alternatywne miejsca składania 3′ i 5′ i wykluczenie eksonu, generując łącznie ponad 500 izoform białka [61,82]. Nieco innym procesem, znacznie mniej poznanym, ale o potencjalnie dużym znaczeniu jest zjawisko transskładania mRNA (trans-splicing). W tym mechanizmie dojrzałe transkrypty są składane z eksonów pochodzących z więcej niż jednego pre-mRNA [28]. Jak dotąd brak jest precyzyjnych danych na temat częstości tego zjawiska, wiadomo natomiast, że jest to proces powszechny wśród organizmów eukariotycznych, także u człowieka [22,26].
Izoformy białek generowane w wyniku alternatywnego składania transkryptu różnią się od siebie strukturą w mniejszym lub większym stopniu. Z różnic ich struktury wynika duża różnorodność funkcjonalna, dzięki czemu izoformy takie pełnić mogą wiele funkcji, odpowiadając za dyskretne różnice w metabolizmie komórki w zależności od jej typu, fazy życiowej czy chwilowych warunków. Oszacowano, że prawie 75% przypadków alternatywnego składania pre-mRNA dotyczy różnic w regionie ulegającym translacji, ma wiec bezpośredni wpływ na strukturę białka [67]. Zmiana właściwości jednej izoformy białka względem drugiej może przyjmować najróżniejsze postaci, zależnie od typu i skali alternatywnego składania, jakie w danym wypadku wystąpiły. W skrajnych przypadkach, przy znacznym skróceniu transkryptu wynikającym z wprowadzenia przedwczesnego kodonu stop lub przesunięciu ramki odczytu, transkrypt taki ulega degradacji w procesie NMD (nonsense-mediated decay), a jak wykazano, proces taki zachodzi w komórce w wyniku alternatywnego składania [44] i uważa się ten proces za element mechanizmu regulacji ekspresji genów [2,14]. Zmiana w strukturze izoformy białka może mieć wpływ na jego zdolność oddziaływania z innymi cząsteczkami, w tym z innymi białkami i kwasami nukleinowymi. Dana izoforma może nie zawierać domeny odpowiedzialnej za wiązanie innej cząstki (np. przez wykluczenie eksonu), czego przykładem może być receptor kwasu retinowego hRXR beta3. Dochodzi tam do utraty zdolności wiązania liganda – kwasu retinowego [51], co jest regulacją negatywną. Ważna jest rola regulacji zdolności oddziaływania z DNA czynników transkrypcyjnych, przez co alternatywne składanie przyczynia się do regulacji ekspresji genów [86]. Zależnie od obecności domeny transbłonowej dana izoforma może być białkiem błonowym lub np. cytoplazmatycznym [1], a brak domen zawierających odpowiednie sygnały lokalizacji w komórce może warunkować ich wewnątrzkomórkową lokalizację, jak w przypadku receptora dopaminy D2 (retencja w retikulum endoplazmatycznym) [74], czy receptora estrogenowego (jądro/cytoplazma) [73]. Także aktywność i inne właściwości enzymów, takie jak powinowactwo, swoistość substratowa czy sposób regulacji mogą się znacznie różnić między izoformami [5]. Nieco innym następstwem alternatywnego składania jest różna stabilność produktu białkowego – czas półtrwania białka może się znacznie różnić dla poszczególnych izoform, np. przez wprowadzenie miejsc trawienia białka [50,76]. Ostatecznie alternatywne składanie w regionach 3′- i 5′ – UTR może wpływać na stabilność mRNA [96].
Efekt fenotypowy alternatywnego składania może się przejawiać na różne sposoby. Mechanizm ten, przez generowanie wysokiego zróżnicowania proteomicznego i ścisłą koordynację alternatywnego składania, zarówno co do miejsca jak i czasu, pozwala na niezwykle precyzyjną regulację procesów komórkowych w zależności od wielu czynników. Przede wszystkim alternatywne składanie ma charakter tkankowoswoisty [16,97]. Oznacza to, że różne izoformy białka mogą być wytwarzane w różnych tkankach w istotnie różniących sie proporcjach [11]. Na tym tle wyróżnić należałoby komórki ośrodkowego układu nerwowego charakteryzujące się szczególnie wyspecjalizowanym zestawem izoform białkowych [20,25]. Innym czynnikiem różnicującym proteom komórki jest zależność od fazy rozwojowej oraz etapu różnicowania komórki [64]. Zidentyfikowano wiele wewnętrznych i zewnętrznych czynników modulujących alternatywne składanie mRNA. Są wśród nich zarówno procesy fizjologiczne, jak aktywacja komórkowego receptora czy zmiany aktywności neuronów, a także procesy będące odpowiedzią na zewnętrzny czynnik, np. zmiana pH, promieniowanie UV lub zmiany warunków osmotycznych [62,93].
Regulacja alternatywnego składania na poziomie molekularnym wydaje się procesem bardzo złożonym, składającym się z wielu współdziałających mechanizmów w znacznej mierze jeszcze niepoznanych (ryc. 3). Klasyczny, najlepiej poznany mechanizm regulacji alternatywnego składania obejmuje wzajemne oddziaływanie elementów cis, na które składają się swoiste sekwencje w mRNA, z elementami trans – grupą białkowych czynników rozpoznających odpowiednie sekwencje w transkrypcie [30]. Elementy cis dzieli się na sekwencje wzmacniające, promujące wycinanie (enhancers) oraz elementy wyciszające, hamujące wycinanie (silencers). Obie te grupy dzieli się z kolei ze względu na lokalizację na intronowe i eksonowe – najlepiej poznane są eksonowe wzmacniacze składania (ESEs – exonic splicing enhancers), wiążące elementy trans – białka bogate w serynę i argininę SR (serine/arginine-rich) [30]. Eksonowe wygaszacze składania (ESSs – exonic splicing silencers) wiążą białka z rodziny hnRNP – białek wchodzących w skład heterogennych rybonukleoprotein, które przyjmują zazwyczaj rolę elementów hamujących trans. Prawdopodobnie czynniki te wiążąc się z sekwencją w eksonie działają bezpośrednio przez współzawodniczenie z czynnikami składania U1 i U2snRNP, aczkolwiek szczegóły tego mechanizmu nie zostały ostatecznie wyjaśnione [30,64]. Rozpatrywane są różne sposoby regulacji, takie jak bezpośrednie blokowanie steryczne elementów składania mRNA, rekrutacja pozytywna, polegająca na promowaniu przyłączania czynników składania w danym miejscu mRNA, czy oddziaływanie z tymi czynnikami i modulowanie ich właściwości [64]. Stwierdzono, że czynniki hnRNP mogą oligomeryzować na nici mRNA i w ten sposób oddziaływać na bardziej odległe fragmenty transkryptu. Uważa się też, że mogą działać przez wypętlanie eksonu, przez co jest on niedostępny dla elementów kompleksu składającego [105]. Elementy intronowe – ISS (intronic splicing silencers) oraz ISE (intronic splicing enhancers) są również obecne, ale są o wiele słabiej poznane [33]. Wśród ISE stosunkowo dobrze scharakteryzowano tzw. tryplety G lub trakt G – powszechne w intronach bogatych w guaninę, a także spotykane w intronach powtórzenia CA, mogące pełnić rolę ISE [105]. Obecność powszechnych elementów cis i regulatorów trans nie tłumaczy w pełni złożoności regulacji alternatywnego składania. Odkrywane są nowe, bardziej swoiste mechanizmy regulacji. Sekwencje cis są w pewnym stopniu zdegenerowane, co przekładać się może na zróżnicowane powinowactwo do elementów trans [33]. Zaproponowano model „ying-yang”, w którym występuje dynamiczna równowaga między oddziaływaniem elementów promujących i hamujących wycinanie, a niewielka przewaga jednego z nich decyduje o tym, czy dany fragment mRNA pozostanie, czy zostanie wycięty [19]. Zidentyfikowane też zostały regulatory tkankowoswoiste, np. NOVA, nPTB, FOX1/2, ESRP1/2 w układzie nerwowym [64,93]. Regulatory takie mogą ulegać modyfikacjom potranslacyjnym, także jako elementy efektorowe dróg sygnalizacji wewnątrzkomórkowej (np. Ras/Raf/MEK/ERK, czy Ca2+/kalmodulina/CamkIV) [33]. Ważna jest też lokalizacja danej sekwencji [105], wzajemne położenie jednej sekwencji względem innych i liczba powtórzeń motywu, co stanowi o sile jej oddziaływania [110]. Interesującym, a zarazem znaczącym w kontekście regulacji alternatywnego składania wydaje się sprzężenie składania mRNA z transkrypcją, „transcriptional coupling”. Składanie odbywa się często (zwłaszcza w przypadku dłuższych transkryptów) kotranskrypcyjnie [70] i jak sie uważa, tempo transkrypcji ma wpływ na wzorzec składania transkryptu [48]. Najnowsze informacje wskazują też na udział domeny CTD polimerazy II RNA w modulacji alternatywnego składania [58].

Ryc. 3. Kontrola alternatywnego składania RNA przez elementy pozytywne i negatywne (na podstawie [105] zmodyfikowano). Wzajemne oddziaływanie elementów trans – białek SR i hnRNP z eksonowymi i intronowymi elementami cis decyduje o wyborze miejsca składania. U1 i U2 – rybonukleoproteiny, składniki kompleksu składania RNA; ESE/ESS – eksonowy wzmacniacz/wygaszacz składania; ISE/ISS – intronowy wzmacniacz/wygaszacz składania
Schorzenia związane z zaburzeniami składania alternatywnego
Bardzo duża złożoność mechanizmu alternatywnego składania pre-mRNA, a zwłaszcza wielopoziomowego systemu jego regulacji pozwala przypuszczać, że jest on bardzo wrażliwy na zaburzenia. Co więcej, zaburzenia alternatywnego składania mogą wywoływać wiele, często trudnych do przewidzenia konsekwencji i prowadzić do rozwoju stanów patologicznych, nierzadko o poważnych następstwach dla całego organizmu. Mnogość nie w pełni jeszcze poznanych sekwencji regulatorowych stanowić może przeszkodę w ustalaniu związku między stanem patologicznym a zaburzeniem w alternatywnym składaniu transkryptu. Mimo to, zidentyfikowano wiele schorzeń o dobrze udokumentowanym związku z procesem alternatywnego składania. Innym stanom chorobowym towarzyszą zmiany wzorca alternatywnego składania wielu genów, trudno jednak jednoznacznie określić, czy jest to następstwem, czy przyczyną procesu chorobowego. Przyczyną stanu patologicznego może być mutacja w genie, którego etiologicznie stan ten dotyczy lub w innym miejscu w genomie, przez co wpływ ten może być pośredni [93]. Proces chorobowy może być następstwem zarówno mutacji w sekwencjach sygnałowych składania, sekwencjach regulatorowych cis, jak też w sekwencjach genów czynników białkowych trans, biorących udział w regulacji składania alternatywnego [93]. Najprostszym i najbardziej oczywistym przykładem mogą być mutacje umiejscowione w trzech podstawowych dla składania sekwencjach najwyższej zgodności – miejsc składania 3′ i 5′ oraz miejsca rozgałęzienia. Takie mutacje w oczywisty sposób zaburzają składanie znosząc oddziaływanie pre-mRNA z czynnikami U1 i U2snRNP. Jak oszacowano, około 9-10% schorzeń genetycznych związanych z mutacjami punktowymi to wynik mutacji w tych trzech sekwencjach [104]. Prawie 15% mutacji punktowych związanych z genetycznymi schorzeniami człowieka dotyczy składania pre-mRNA [33], a 60% z nich związane jest z mutacjami w obszarach końcowych eksonów 3′ i 5′, co może utrudniać ich rozpoznanie [65]. Poza wymienionymi sekwencjami najwyższej zgodności eksony zawierać mogą bardzo różnorodne sekwencje regulatorowe o charakterze wzmacniającym lub wyciszającym składanie. Jak się ocenia, prawie 25% mutacji synonimicznych w eksonach istotnie wpływa na proces składania, zaburzając jego prawidłowy wzorzec [69]. Dla niektórych genów nawet ponad 50% mutacji punktowych w ich eksonach może istotnie zaburzać prawidłowe składanie [46]. Cząsteczki mRNA generowane w wyniku prawidłowego alternatywnego składania ulegają procesowi translacji dając w pełni funkcjonalne białko. Także część izoform nieprawidłowych, generowanych w wyniku zaburzeń w składaniu pre-mRNA może ulegać translacji do białka o mniej lub bardziej zmienionej funkcji względem białka prawidłowego. Pozostałe, nieprawidłowe transkrypty mające przedwczesny kodon stop, ulegać mogą procesowi degradacji przez mechanizm NMD (nonsense-mediated decay), inne niezawierające kodonu stop – procesowi NSD (nonstop mRNA decay) lub NGD (no-go decay), jeśli z powodu błędnej budowy powodują zatrzymanie procesu translacji [31,34]. Rodzinna dysautonomia, FD (familial dysautonomia) jest przykładem schorzenia związanego z wprowadzeniem przez alternatywne składanie przedwczesnego kodonu STOP. Jest to neuropatia czuciowa dziedziczona recesywnie spowodowana mutacją w genie IKBKAP, którego produkt funkcjonuje jako czynnik transkrypcyjny [106]. Synonimiczna tranzycja T>C w ostatnim nukleotydzie 20 intronu po stronie 5′ miejsca wycinania powoduje pominięcie 20 eksonu, co przesuwa ramkę odczytu za tym miejscem i wprowadza przedwczesny kodon STOP. Taki skrócony wariant białka częściowo ulega degradacji w procesie NMD, przez co zostaje ograniczona pula tego białka w komórce [106].
Innym, charakterystycznym przykładem zespołu genetycznego powiązanego z zaburzeniami w alternatywnym składaniu jest dystrofia mięśniowa Duchenne’a – najczęściej występująca postać dystrofii mięśniowej sprzężona z płcią [66]. Jest to schorzenie o zróżnicowanej patogenezie molekularnej i choć większość przypadków choroby wiąże się z delecją w genie dystrofiny, składającego się z 79 eksonów, to również znaczna część przypadków choroby wiąże się z występowaniem punktowych mutacji zarówno w eksonach jak i intronach, w wyniku czego zaburzone zostaje składanie [66]. Przykładem takiej mutacji jest zmiana T>A w eksonie 31, wprowadzająca przedwczesny kodon STOP, ale jednocześnie wprowadza nowe miejsce ESS wiążące negatywny cis-regulator hnRNPA1, przez co ekson ten jest częściowo pomijany [13]. Takie białko zachowuje częściową funkcjonalność, dając łagodniejszy w przebiegu obraz choroby [13].
Zaburzenie alternatywnego składania może mieć wymiar ilościowy. Różne mutacje w DNA mogą prowadzić do takiego samego lub też podobnego efektu – mogą zmieniać proporcje izoform prawidłowych i nieprawidłowych, lecz w różnym stopniu. Na przykład mutacja G>A pierwszego nukleotydu eksonu 6 w genie ATP7A – białka uczestniczącego w metabolizmie miedzi w organizmie człowieka, prowadzi do pominięcia tego eksonu, czego następstwem jest ostra postać schorzenia neurodegeneracyjnego związanego z upośledzoną zdolnością asymilacji miedzi – choroby Menkesa [7]. Jednocześnie inna mutacja, podstawienie U>A w pozycji +6 eskonu 6 skutkuje częściowym tylko wykluczeniem prawidłowej izoformy produktu ATP7A, a to w konsekwencji prowadzi do schorzenia o łagodniejszym przebiegu – tzw. zespołu rogu potylicznego (occipital horn syndrome), charakteryzującego się głównie istnieniem zmian kostnych [57].
Tauopatie, to grupa schorzeń o charakterze neurodegeneracyjnym, związana z defektami w alternatywnym składaniu białka tau. Rola tego białka ograniczona jest w znacznej mierze do komórek układu nerwowego, jest kodowane przez gen MAPT (microtubule associated protein tau) i na skutek alternatywnego składania generowanych jest wiele jego izoform – 8 z 16 jego eksonów ulega alternatywnemu składaniu [98]. Dodatkowo, alternatywne składanie tego białka jest ściśle zależne zarówno od fazy rozwoju osobniczego, jak też od ściśle zdefiniowanego umiejscowienia komórek w obrębie mózgu. Białko tau wiąże mikrotubule dzięki obecności powtórzeń regionów wiązania tych białek. Zidentyfikowano wiele mutacji wpływających na alternatywne składanie tego białka i jednocześnie ich związek z występowaniem takich schorzeń, jak choroba Alzheimera czy otępienie skroniowo-czołowe [98]. Mutacje dotyczące otępienia skroniowo-czołowego (zidentyfikowano 42 takie mutacje) dotyczą w głównej mierze eksonu 10. Bardziej szczegółowe badania sugerują, że proces chorobowy może być w tym przypadku wynikiem zmian wzajemnych proporcji poszczególnych izoform białka tau w wyniku zaburzających alternatywne składanie mutacji, a nie zmiany, czy utraty funkcji przez te izoformy [12,98].
Osobną grupą schorzeń o potwierdzonym związku z zaburzeniami w alternatywnym składaniu transkryptu są nowotwory złośliwe. Mutacje w genach prowadzące do zmian we wzorcu alternatywnego składania mogą być związane z transformacją nowotworową. Jednym z przykładów może być gen LKB1, supresor nowotworowy, kodujący kinazę serynowo-treoninową biorącą udział w kontroli apoptozy i cyklu komórkowego. Mutacja w tym genie związana jest z syndromem Peutza-Jeghersa, objawiającego się polipowatością jelita i dużą predyspozycją do nowotworów. W wyniku mutacji powstaje alternatywne miejsce składania 3′, a w konsekwencji dochodzi do przesunięcia ramki odczytu i wprowadzenia przedwczesnego kodonu STOP [24]. Innym przykładem może być gen BRCA1 – dobrze poznany supresor nowotworowy, którego mutacje związane są z dużym ryzykiem raka piersi. Jedna z mutacji w tym genie – mutacja nonsensowa w eksonie 18 – wpływa na zaburzenie alternatywnego składania przez zniesienie miejsca ESE (dotąd zidentyfikowano 23 sekwencje ESE w tym genie) i powoduje pominięcie eksonu 18 [92]. Ponadto w wielu nowotworach wykryto istotne zmiany we wzorcu alternatywnego składania wielu genów, a nie zostały znalezione mutacje w ich sekwencji mogące leżeć u podstaw takich zmian. Poza tym, niektóre izoformy produktów tych genów ulegają kumulacji w rozwoju guza, a potwierdzono też, że sztuczna nadekspresja tych wariantów może powodować transformację nowotworową komórki [88,92]. Pozwala to przypuszczać, że mutacje te mogą dotyczyć genów kodujących elementy regulatorowe trans [92]. Za ciekawy przykład może tu posłużyć dobrze poznany supresor nowotworowy p53. Białko to może występować pod postacią kilku izoform [68], a ich występowanie jest tkankowoswoiste, a także proporcje ich są zaburzone w nowotworze piersi [9]. Alternatywnemu składaniu ulegają też główne regulatory białka p53, w tym MDM2. Zidentyfikowano ponad 40 alternatywnych transkryptów tego białka, z czego część uważa się za cząsteczki nieprawidłowe, związane z nowotworzeniem [92]. Podkreśla to rolę, jaką w procesie nowotworzenia odgrywać może alternatywne składanie transkryptów i wskazuje na ważny kierunek badań nad biologią molekularną nowotworów, a jednocześnie otwiera drogę do nowych potencjalnych terapii chorób nowotworowych.
Interferencja RNA w terapii chorób związanych z alternatywnym składaniem
Schorzenia będące wynikiem błędnego alternatywnego składania mają przeważnie ciężki przebieg, co więcej leczenie tych schorzeń tradycyjnymi metodami jest problematyczne, a w wielu przypadkach niemożliwe, co nadaje im status nieuleczalnych i często prowadzą do śmierci chorego. Z tego powodu poszukiwanie nowych, skutecznych metod terapii takich schorzeń znajduje się w centrum zainteresowania wielu grup badawczych. Interferencja RNA (RNAi – RNA interference) jest zachowanym ewolucyjnie mechanizmem regulacji ekspresji genów u eukariontów (ryc. 4). Należy do grupy zależnych od RNA mechanizmów wyciszania genów. Umożliwia degradację swoistych cząsteczek mRNA, bądź też zablokowanie translacji, zależnie od ich sekwencji [81]. Najlepiej i najwcześniej poznanym jest udział w interferencji cząsteczek siRNA (short interferring RNA). Powstają one z dłuższych, dwuniciowych cząsteczek prekursorowych, ulegających cięciu przez rybonukleazę III, Dicer, do 21-25-nukleotydowych dwuniciowych cząsteczek, które wchodzą następnie w skład dużych rybonukleoproteinowych kompleksów RISC (RNA-induced silencing complex), gdzie zostają rozwinięte do form jednoniciowych [81]. Jako składnik kompleksu RISC siRNA uczestniczą w rozpoznawaniu i degradacji komplementarnych do nich cząsteczek mRNA. Za degradację docelowych mRNA odpowiada obecna w kompleksie RISC endonukleaza Argonaute 2 (Ago-2) z rodziny białek Argonaute [45]. Nieco odmiennym typem cząsteczek RNA biorącym udział w interferencji jest miRNA (microRNA). Wyciszanie genów zależne od miRNA może się odbywać zarówno przez degradację docelowych RNA (podobnie jak siRNA), jak też przez zablokowanie translacji mRNA (częściej) [3]. Podobnie jak siRNA, miRNA powstają z większych prekursorów poprzez działanie rybonukleazy Dicer [99]. Biogeneza miRNA jest procesem wieloetapowym. Pierwotne prekursory – pri-miRNA, często pochodzące z intronów genów transkrybowanych przez polimerazę RNA II, podlegają obróbce przez rybonukleazę Drosha. Daje to ~70-nukleotydowy produkt pośredni o strukturze szpilki do włosów [42], który jest następnie transportowany do cytoplazmy, gdzie po ostatecznej obróbce przez białko Dicer powstaje gotowy produkt – miRNA. Dojrzałe cząsteczki miRNA wchodzą w skład kompleksów rybonukleoproteinowych miRNP i na tym etapie mogą uczestniczyć w interferencji RNA [6].

Ryc. 4. Mechanizm interferencji RNA z udziałem siRNA. Prekursor siRNA (dsRNA) podlega cięciu przez kompleks DICER, generując dwuniciowe siRNA. SiRNA wchodzi w skład kompleksu RISC, w wyniku rozplatania siRNA do postaci jednoniciowej następuje aktywacja RISC. Aktywny kompleks RISK rozpoznaje docelową cząsteczkę mRNA indukując jej degradację
Mimo iż proces interferencji RNA odkryto stosunkowo niedawno [18], zaproponowano wiele jego potencjalnych zastosowań, a wiele jest obecnie w powszechnym użyciu, zarówno jako narzędzie terapeutyczne, jak i badawcze. Rutynowo stosuje sie je w badaniach nad funkcją genów, poprzez kontrolowane ich wyciszanie [47]. Koncepcję zastosowania interferencji RNA do korekty zaburzeń alternatywnego składania (ryc. 5) można uznać za względnie nowy punkt widzenia, z czego wynika niewielka, jak dotąd liczba spektakularnych sukcesów. Jednak wiele opublikowanych wyników pozwala optymistycznie oceniać rozwój badań w tym kierunku i potwierdza słuszność rozwijania metod opierających się na interferencji RNA. Przede wszystkim jednak podejście to daje nadzieję na eliminację wielu problemów spotykanych przy rozwoju wcześniejszych, standardowych technik molekularno-genetycznych opierających się na terapii genowej, takie jak niewielka efektywność transferu genu, znaczne ograniczenie w wielkości wprowadzanego fragmentu genu, negatywne efekty zależne od miejsca wprowadzenia transgenu, czy reakcja obronna komórki i organizmu związana z procesem transfekcji [108]. Podjęto już pierwsze próby zastosowania interferencji RNA zarówno w badaniach na komórkach, jak również w leczeniu schorzeń u człowieka. Osiągnięto też pierwsze sukcesy, czego przykładem może być próba zastosowania interferencji RNA w leczeniu wysiękowej postaci zwyrodnienia plamki związanego z wiekiem (AMD – age-related macular degeneration), z zastosowaniem siRNA w celu wyciszenia genu receptora śródbłonkowego czynnika wzrostu naczyń, (VEGFR1 – vascular endothelial growth factor receptor 1) [54], jednego z czynników odpowiedzialnych za patologiczny rozrost choriokapilar w siatkówce w wysiękowej postaci AMD. Inny zespół raportował, że w pierwszej fazie badań klinicznych iniekcja do gałki ocznej siRNA komplementarnego do transkryptu genu VEGFR1 (Sirna-027) skutkowało zatrzymaniem progresji choroby, a często także poprawą ostrości widzenia w grupie badanej. Nie obserwowano również działania toksycznego podanej substancji [34].

Ryc. 5. Korekta alternatywnego składania przez interferencję RNA, powstają dwa transkrypty różniące się fragmentem (prawidłowy – zielona wstawka i nieprawidłowy – czerwona wstawka). Wprowadzone do komórki siRNA współtworzy aktywny kompleks RISC, rozpoznaje nieprawidłowy wariant i go degraduje. Prawidłowy wariant nie zostaje rozpoznany i nie ulega degradacji
Projektuje się cząsteczki siRNA swoiste dla określonych, wytwarzanych w komórce transkryptów, wyciszając w ten sposób określone geny, których zaburzona ekspresja jest często charakterystycznym symptomem w stanach chorobowych [23]. Liczne próby podjęto w terapii nowotworów, ze względu na nie tylko oporność na tradycyjne schematy terapeutyczne i dużą śmiertelność, ale i ze względu na swoiste, odmienne od stanu prawidłowego, wzorce ekspresji wielu genów w nowotworach, z obniżonym poziomem ekspresji grupy genów przy jednoczesnej nadekspresji innych [3]. Dotyczy to zwłaszcza genów związanych z regulacją takich procesów jak apoptoza, neowaskularyzacja, czy podziały komórki. Ponadto u podstaw procesu nowotworzenia leżą kaskady następujących po sobie mutacji w obrębie głównych genów [3], a przez to genów wydających się właściwym celem terapii z użyciem interferującego RNA. Liczne próby zastosowania interferencji RNA w terapii związane były ze swoistą dla odpowiedzi na infekcje wirusowe reakcją przeciw wprowadzanym do komórki syntetycznym dwuniciowym RNA. W tym celu stosowano transfekcję komórek krótkimi jednoniciowymi cząsteczkami RNA zawierającymi dwunukleotydowe jednoniciowe końce 3′, co znosiło negatywne działanie związane z reakcją obronną komórki [15]. Wkrótce też uzyskano ekspresję cząsteczki zawierającej promotor polimerazy RNA III krótkich cząsteczek RNA o strukturze szpilki do włosów – shRNA (short hairspin RNA), naśladujących miRNA [21]. Takie cząsteczki shRNA ulegają następnie obróbce w komórce przez RNAzę III, po czym zostają włączone w skład kompleksu RISC [53].
Wraz z rozwojem technik opartych na interferencji RNA, jak i próbami ich zastosowań klinicznych rozwinął się kierunek badań w biologii molekularnej, o znacznym potencjale i dużej precyzji w kontroli ekspresji genów. O ile jednak zablokowanie funkcji poszczególnych genów nie stanowi obecnie dużego wyzwania, to próby wyciszenia poszczególnych alleli czy izoform jest podejściem o wiele bardziej problematycznym (ryc. 6). Wyciszenie izoformy wyróżniającej się obecnością lub brakiem pojedynczego eksonu, bądź też zawierającej skrócony/wydłużony ekson w stosunku do izoformy prawidłowej stanowi techniczny problem ze względu na duże podobieństwo w sekwencji izoform i duże ryzyko ich jednoczesnego wyciszenia. Co więcej, o ile względnie łatwym zadaniem jest ukierunkowanie interferencji RNA na alternatywny ekson, znacznie trudniejszym zadaniem jest zorientowanie interferującego RNA na granicę ekson-intron. Problemem jest tu znaczne podobieństwo sekwencji w ściśle określonym miejscu stanowiącym zarazem granicę między eksonem i intronem [21]. Niedostateczna precyzja w wyciszaniu izoformy białka może spowodować całkowite zahamowanie ekspresji genu (wyeliminowanie także innych, poza docelową, prawidłowych izoform białka), a taka ingerencja może zakończyć się śmiercią komórki [21].
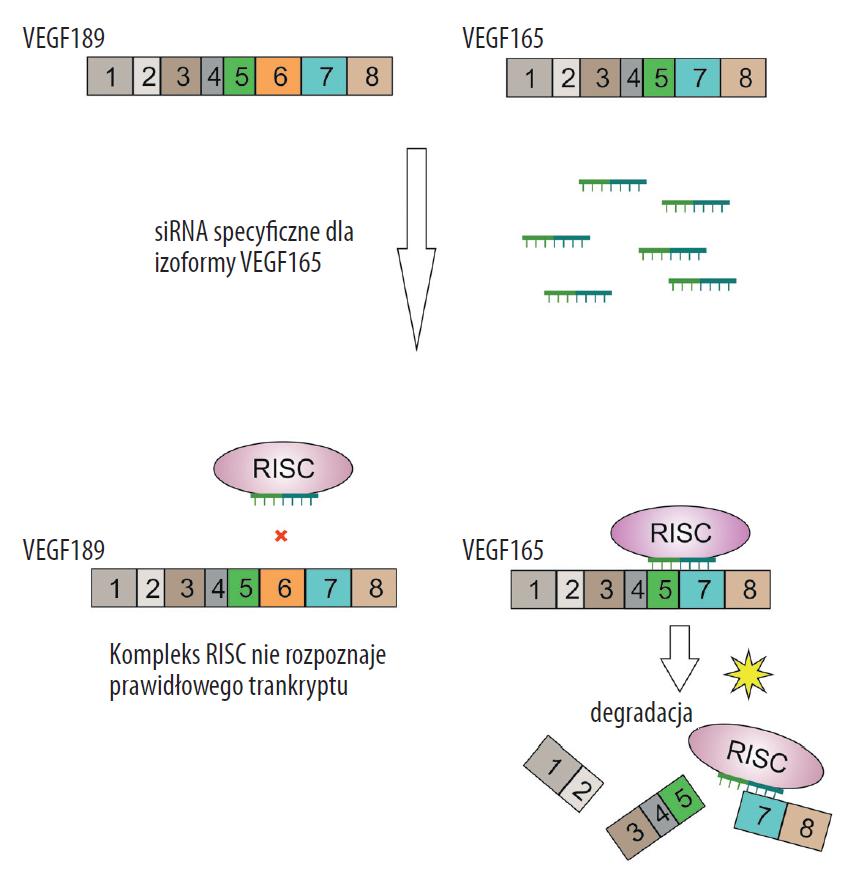
Ryc. 6. Zastosowanie interferencji RNA w wyciszaniu izoformy białka VEGF jako celu terapii nowotworów (na podstawie [108] zmodyfikowano). Izoforma VEGF165 nie ma eksonu 6 i ulega wyciszaniu w procesie interferencji RNA. VEGF189 – prawidłowa cząsteczka mRNA, VEGF165 – mRNA z brakującym eksonem 6; kolorowe prostokąty odpowiadają poszczególnym eksonom mRNA
W terapiach antynowotworowych jednym z głównych i oczywistych potencjalnych celów interferencji RNA wydają się geny biorące udział w regulacji procesu apoptozy. W komórkach nowotworowych zachodzi wiele zmian we wzorcu ekspresji genów, w tym genów związanych z regulacją apoptozy, wzrostu i różnicowania komórek. Dotyczy to zarówno poziomu ekspresji tych genów, jak i względnych poziomów poszczególnych ich izoform. Alternatywne składanie mRNA jest ważnym punktem w regulacji ekspresji tych genów, ich ekspresja wykazuje w wielu przypadkach silny swoisty tkankowo charakter i jak wiadomo, wiąże się z występowaniem poszczególnych typów nowotworów [21]. Wśród białek z rodziny Bcl-2, o ważnej funkcji w regulacji apoptozy, produkt genu Bcl-x ulega alternatywnemu składaniu, w wyniku czego powstają dwie izoformy o przeciwstawnym działaniu – Bcl-xL o właściwościach antyapoptotycznych oraz Bcl-xS, którego przewaga w komórce promuje apoptozę [8]. Wyciszenie izoformy Bcl-xL uwrażliwia komórki nowotworowe na indukowaną chemioterapeutykami apoptozę [111,112]. Podobnie błonowy receptor Fas jest pozytywnym elementem sygnalizacji w apoptozie, jednakże występująca w mniejszości izoforma tego białka, pochodząca z transkryptu pozbawionego eksonu 6, będąca cytoplazmatycznym wariantem receptora Fas nie ma zdolności indukowania apoptozy [107]. Inaktywacja tej izoformy receptora Fas może zatem zwiększać wrażliwość komórki na sygnały proapoptotyczne [21]. Nieco innym przykładem jest supresor nowotworowy – białko KLF6, wykazujący dysfunkcję w nowotworze prostaty [10]. Związana z tym nowotworem mutacja IVSΔA w genie KLF6 skutkuje nadmiernym wytwarzaniem izoformy SV1 względem pozostałych izoform – SV2 i SV3. Izoformy SV2 i SV3 działają antyproliferacyjnie, z kolei izoforma SV1 stymuluje podziały komórkowe [59]. SiRNA skierowane przeciwko izoformie SV1 produktu genu KLF6 ograniczało wzrost guza o 50% i dodatkowo powodowało spadek ekspresji innych, związanych ze wzrostem guza i angiogenezą genów [60], okazując się przez to obiecującą metodą w terapii nowotworu prostaty.
Prawie 30% przypadków nowotworów piersi i jajnika jest związanych z nadekspresją receptora estrogenowego HER-2, co oznacza gorsze rokowania dla pacjenta [89]. Wyciszanie genu HER-2 za pomocą interferencji RNA w przypadkach HER-2-zależnych nowotworów wyraźnie ogranicza proliferację komórek guza i promuje ich apoptozę [17]. Co więcej, produkt genu HER-2 ulega alternatywnemu składaniu, a obecność niedawno zidentyfikowanej izoformy DHER-2 może się wiązać z większą aktywnością transformacyjną komórek, a przez to wydaje się potencjalnie wartościowym celem terapii za pomocą interferencji RNA ukierunkowanej na konkretną izoformę białka [40].
Trwają także intensywne prace nad zastosowaniem interferencji RNA w schorzeniach neurodegeneracyjnych. Dobrze scharakteryzowana została grupa mutacji w genie tau, leżąca u podstaw wielu schorzeń w obrębie układu nerwowego, w tym demencja czołowo-skroniowa i parkinsonizm, czy postępujące porażenie nadjądrowe [29]. Mutacja V337M wewnątrz eksonu 12 związana jest z demencją czołowo-skroniową. Ponieważ jest to mutacja punktowa, dużym wyzwaniem było skonstruowanie takiej cząsteczki siRNA, która charakteryzowałaby się selektywnością w stosunku do wadliwego wariantu, nie wpływając na ekspresję wariantu prawidłowego [55].
Szczególnie istotne z punktu widzenia terapii skierowanej na proces alternatywnego składania RNA są zaburzenia związane z mutacjami w genach czynników uczestniczących w samym procesie składania mRNA. Klasycznym tego przykładem są mutacje w genach SMN1 i SMN2, które leżą u podstaw rdzeniowej atrofii mięśni (SMA – spinal muscular athrophy) [80]. Produkty tych genów ulegają konstytutywnej ekspresji w różnego rodzaju tkankach i pełnią istotne funkcje na etapie montażu kompleksu rybonukleoproteinowego składania mRNA [4]. W następstwie substytucji C>T w pozycji 6 eksonu 7 genu SMN2 dochodzi do nieprawidłowego alternatywnego składania transkryptu, w wyniku czego w komórce pojawia się przewaga nieprawidłowej izoformy pozbawionej 7 eksonu [43]. Większość typów komórek jest w stanie skompensować lub tolerować ten defekt, jednakże w przypadku motoneuronów dochodzi do ich obumierania [4]. Molekularno-genetyczny mechanizm tego zjawiska nie został jeszcze wyjaśniony, a rozważane są dwa modele tłumaczące to schorzenie. Pierwszy model zakłada istnienie eksonowego wzmacniacza składania w eksonie 7, a mutacja w jego obrębie znosi skutki wzmacniania [36]. W alternatywnym modelu mutacja w eksonie 7 powoduje powstanie eksonowego wygaszacza składania, który przez przyłączenie cząsteczki hnRNP A1 promuje usuwanie tego eksonu [36]. Przeprowadzono próbę korekcji polegającą na wyciszeniu genu czynnika hnRNP A1 (a także pokrewnego mu czynnika hnRNP A2), za pomocą interferencji RNA. W wyniku takiego działania udało się przywrócić prawidłowy fenotyp, a ekspresja wadliwej izoformy SMN2 została wygaszona na rzecz pełnego, funkcjonalnego białka, zawierającego domenę kodowaną w eksonie 7 [36].
Przedstawione informacje mają na celu przedstawienie obecnego stanu wiedzy i rozwoju metod terapeutycznych z użyciem procesu interferencji RNA. Dzięki intensywnym próbom nad stosowaniem interferencji RNA w terapii, metoda ta jest na etapie dynamicznego rozwoju, obejmując coraz liczniejszą grupę schorzeń, jako potencjalnych celów w terapii. Nie jest wykluczone, że udział alternatywnego składania w patogenezie okaże się istotnym elementem większej liczby chorób, w których aspekt ten nie został jeszcze zbadany. W tym kontekście wyciszanie wadliwych wariantów białek wydaje się podejściem znacznie bardziej precyzyjnym niż standardowe terapie i jednocześnie dającym ogromne możliwości i nadzieję na skuteczne leczenie dużej grupy schorzeń będących wciąż chorobami nieuleczalnymi. Pomijając aspekty techniczne stanowiące pewne ograniczenia w korekcji składania mRNA, jedynym problemem zdaje się pozostawać znajomość molekularnych podstaw w patogenezie schorzeń człowieka, których poznanie pozwala na wyznaczenie molekularnych celów terapii, a w przypadku terapii opartej na interferencji RNA daje nadzieje na opracowanie niezwykle skutecznych i nieosiągalnych dotychczas schematów leczenia.
PIŚMIENNICTWO
[1] Alt F.W., Bothwell A.L., Knapp M., Siden E., Mather E., Koshland M., Baltimore D.: Synthesis of secreted and membrane-bound immunoglobulin mu heavy chains is directed by mRNAs that differ at their 3’ends. Cell, 1980; 20: 293-301
[PubMed]
[2] Amor S., Remy S., Dambrine G., Le Vern Y., Rasschaert D., Laurent S.: Alternative splicing and nonsense-mediated decay regulate telomerase reverse transcriptase (TERT) expression during virus-induced lymphomagenesis in vivo. BMC Cancer, 2010; 10: 571
[PubMed] [Full Text HTML] [Full Text PDF]
[3] Angaji S.A., Hedayati S.S., Poor R.H., Madani S., Poor S.S., Panahi S.: Application of RNA interference in treating human diseases. J. Genet., 2010; 89: 527-537
[PubMed] [Full Text PDF]
[4] Bebee T.W., Gladman J.T., Chandler D.S.: Splicing regulation of the survival motor neuron genes and implications for treatment of spinal muscular atrophy. Front. Biosci., 2010; 15: 1191-1204
[PubMed] [Full Text HTML] [Full Text PDF]
[5] Benedict C.L., Gilfillan S., Kearney J.F.: The long isoform of terminal deoxynucleotidyl transferase enters the nucleus and, rather than catalysing nontemplated nucleotide addition, modulates the catalytic activity of the short isoform. J. Exp. Med., 2001; 193: 89-99
[PubMed] [Full Text HTML] [Full Text PDF]
[6] Bernstein E., Caudy A.A., Hammond S.M., Hannon G.J.: Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 2001; 409: 363-366
[PubMed]
[7] Bertini I., Rosato A.: Menkes disease. Cell Mol. Life Sci., 2008; 65: 89-91
[PubMed]
[8] Boise L.H., Gonzalez-Garcia C.E., Postema C.E., Ding L., Lindsten T., Turka L.A., Mao X., Nunez G., Thomson C.B.: Bcl-x, a Bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell, 1993; 74: 597-608
[PubMed]
[9] Bourdon J.C., Fernandes K., Murray-Zmijewski F., Liu G., Diot A., Xirodimas D.P., Saville M.K., Lane D.P.: p53 isoforms can regulate p53 transcriptional activity. Genes Dev., 2005; 19: 2122-2137
[PubMed] [Full Text HTML] [Full Text PDF]
[10] Chen C., Hyytinen E.R., Sun X., Helin H.J., Koivisto P.A., Frierson H.F.Jr, Vessella R.L., Dong J.T.: Deletion, mutation and loss of expression of KLF6 in human prostate cancer. Am. J. Pathol., 2003; 162: 1349-1354
[PubMed] [Full Text HTML] [Full Text PDF]
[11] Chen Z., Gao X., Lei T., Chen X., Zhou L., Yu A., Lei P., Zhang R., Long H., Yang Z.: Molecular characterization, expression and chromosomal localization of porcine PNPLA3 and PNPLA4. Biotechnol. Lett., 2011; 33: 1327-1337
[PubMed]
[12] Dawson H.N., Cantillana V., Chen L., Vitek M.P.: The tau N279K exon 10 splicing mutation recapitulates frontotemporal dementia and parkinsonism linked to chromosome 17 tauopathy in a mouse model. J. Neurosci., 2007; 27: 9155-9168
[PubMed] [Full Text HTML] [Full Text PDF]
[13] Disset A., Bourgeois C.F., Benmalek N., Claustres M., Stevenin J., Tuffery-Giraud S.: An exon skipping-associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements. Hum. Mol. Genet., 2006; 15: 999-1013
[PubMed] [Full Text HTML]
[14] Durand C., Roeth R., Dweep H., Vlatkovic I., Decker E., Schneider K.U., Rappold G.: Alternative splicing and nonsense-mediated RNA decay contribute to the regulation of SHOX expression. PLoS One, 2011; 6: e18115
[PubMed] [Full Text HTML] [Full Text PDF]
[15] Elbashir S.M., Lendeckel W., Tuschl T.: RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev., 2001; 15: 188-200
[PubMed] [Full Text HTML] [Full Text PDF]
[16] Emig D., Albrecht M.: Tissue-specific proteins and functional implications. J. Proteome Res., 2011; 10: 1893-1903
[PubMed] [Full Text HTML] [Full Text PDF]
[17] Faltus T., Yuan J., Zimmer B., Kramer A., Loibl S., Kaufmann M., Strebhardt K.: Silencing of the HER2/neu gene by siRNA inhibits proliferation and induces apoptosis in HER2/neu-overexression breast cancer cells. Neoplasia, 2004; 6: 786-795
[PubMed] [Full Text HTML] [Full Text PDF]
[18] Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C.: Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 1998; 391: 806-811
[PubMed] [Full Text HTML] [Full Text PDF]
[19] Fu X.D.: Towards a splicing code. Cell, 2004; 119: 736-738
[PubMed] [Full Text HTML] [Full Text PDF]
[20] Gao F.B.: Molecular and cellular mechanisms of dendritic morphogenesis. Curr. Opin. Neurobiol., 2007; 17: 525-532
[PubMed] [Full Text HTML] [Full Text PDF]
[21] Gaur R.K.: RNA interference: a potential therapeutic tool for silencing splice isoforms linked to human diseases. Biotechniques , 2006; Suppl.: 15-22
[PubMed] [Full Text HTML] [Full Text PDF]
[22] Glanz S., Kuck U.: Trans-splicing of organelle introns – a detour to continuous RNAs. Bioessays, 2009; 31: 921-934
[PubMed]
[23] Gupta P.K.: RNA interference-gene silencing by double-stranded RNA: The 2006 Nobel prize for physiology or medicie. Curr. Sci., 2006; 91: 1443
[24] Hastings M.L., Resta N., Traum D., Stella A., Guanti G., Krainer A.R.: An LKB1 AT-AC intron mutation causes Peutz-Jeghers syndrome via splicing at noncanonical cryptic sites. Nat. Struct. Mol. Biol., 2005; 12: 54-59
[PubMed]
[25] Hattori D., Millard S.S., Wojtowicz W.M., Zipursky S.L.: Dscam-mediated cell recognition regulates neural circuit formation. Annu. Rev. Cell Dev. Biol., 2008; 24: 597-620
[PubMed] [Full Text HTML] [Full Text PDF]
[26] Herai R.H., Yamagishi M.E.: Detection of human interchromosomal trans-splicing in sequence databanks. Brief Bioinform., 2010; 11: 198-209
[PubMed] [Full Text HTML] [Full Text PDF]
[27] Hertel K.J.: Combinatorial control of exon recognition. J. Biol. Chem., 2008; 283: 1211-1215
[PubMed] [Full Text HTML] [Full Text PDF]
[28] Horiuchi T., Aigaki T.: Alternative trans-splicing: a novel mode of pre-mRNA processing. Biol. Cell, 2006; 98: 135-140
[PubMed]
[29] Houlden H., Baker M., Morris H.R., MacDonald N., Pickering-Brown S., Adamson J., Lees A.J., Rossor M.N., Quinn N.P., Kertesz A., Khan M.N., Hardy J., Lantos P.L., St George-Hyslop P., Munoz D.G., Mann D., Lang A.E., Bergeron C., Bigio E.H., Litvan I., Bhatia K.P., Dickson D., Wood N.W., Hutton M.: Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology, 2001; 56: 1702-1706
[PubMed]
[30] House A.E., Lynch K.W.: Regulation of alternative splicing: more than just the ABCs. J. Biol. Chem., 2008; 283: 1217-1221
[PubMed] [Full Text HTML] [Full Text PDF]
[31] Hsu S.N., Hertel K.J.: Spliceosomes walk the line: splicing errors and their impact on cellular function. RNA Biol., 2009; 6: 526-530
[PubMed] [Full Text HTML] [Full Text PDF]
[32] Hughes T.A.: Regulation of gene expression by alternative untranslated regions. Trends Genet., 2006; 22: 119-122
[PubMed]
[33] Hui J.: Regulation of mammalian pre-mRNA splicing. Sci. China C. Life Sci., 2009; 52: 253-60
[PubMed]
[34] Isken O., Maquat L.E.: Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev., 2007; 21: 1833-1856
[PubMed] [Full Text HTML] [Full Text PDF]
[35] Kaiser P.K., Symons R.C., Shah S.M., Quinlan E.J., Tabandeh H., Do D.V., Reisen G., Lockridge J.A., Short B., Guerciolini R., Nguyen Q.D.: Sirna-027 Study Investigators.: RNAi-based treatment for neovascular age-related macular degeneration by Sirna-027. Am. J. Ophthalmol., 2010; 150: 33-39.e2
[PubMed]
[36] Kashima T., Manley J.L.: A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet., 2003; 34: 460-463
[PubMed]
[37] Keating K.S., Toor N., Perlman P.S., Pyle A.M.: A structural analysis of the group II intron active site and implication for the spliceosome. RNA, 2010; 16: 1-9
[PubMed] [Full Text HTML] [Full Text PDF]
[38] Kim E., Goren A., Ast G.: Alternative splicing: current perspectives. Bioessays, 2008; 30: 38-47
[PubMed]
[39] Kornblihtt A.R., de la Mata M., Fededa J.P., Munoz M.J., Noques G.: Multiple links between transcription and splicing. RNA, 2004; 10: 1489-1498
[PubMed] [Full Text HTML] [Full Text PDF]
[40] Kwong K.Y., Hung M.C.: A novel splice variant of HER2 with increased transformation activity. Mol. Carcinog., 1998; 23: 62-68
[PubMed]
[41] Lander E.S., Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., Kann L., Lehoczky J., LeVine R., McEwan P., McKernan K., Meldrim J., Mesirov J.P., Miranda C., Morris W., Naylor J., Raymond C., Rosetti M., Santos R., Sheridan A., Sougnez C., Stange-Thomann N., Stojanovic N., Subramanian A., Wyman D., Rogers J., Sulston J., Ainscough R., Beck S., Bentley D., Burton J., Clee C., Carter N., Coulson A., Deadman R., Deloukas P., Dunham A., Dunham I., Durbin R., French L., Grafham D., Gregory S., Hubbard T., Humphray S., Hunt A., Jones M., Lloyd C., McMurray A., Matthews L., Mercer S., Milne S., Mullikin J.C., Mungall A., Plumb R., Ross M., Shownkeen R., Sims S., Waterston R.H., Wilson R.K., Hillier L.W., McPherson J.D., Marra M.A., Mardis E.R., Fulton L.A., Chinwalla A.T., Pepin K.H., Gish W.R., Chissoe S.L., Wendl M.C., Delehaunty K.D., Miner T.L., Delehaunty A., Kramer J.B., Cook L.L., Fulton R.S., Johnson D.L., Minx P.J., Clifton S.W., Hawkins T., Branscomb E., Predki P., Richardson P., Wenning S., Slezak T., Doggett N., Cheng J.F., Olsen A., Lucas S., Elkin C., Uberbacher E., Frazier M., Gibbs R.A., Muzny D.M., Scherer S.E., Bouck J.B., Sodergren E.J., Worley K.C., Rives C.M., Gorrell J.H., Metzker M.L., Naylor S.L., Kucherlapati R.S., Nelson D.L., Weinstock G.M., Sakaki Y., Fujiyama A., Hattori M., Yada T., Toyoda A., Itoh T., Kawagoe C., Watanabe H., Totoki Y., Taylor T., Weissenbach J., Heilig R., Saurin W., Artiguenave F., Brottier P., Bruls T., Pelletier E., Robert C., Wincker P., Smith D.R., Doucette-Stamm L., Rubenfield M., Weinstock K., Lee H.M., Dubois J., Rosenthal A., Platzer M., Nyakatura G., Taudien S., Rump A., Yang H., Yu J., Wang J., Huang G., Gu J., Hood L., Rowen L., Madan A., Qin S., Davis R.W., Federspiel N.A., Abola A.P., Proctor M.J., Myers R.M., Schmutz J., Dickson M., Grimwood J., Cox D.R., Olson M.V., Kaul R., Raymond C., Shimizu N., Kawasaki K., Minoshima S., Evans G.A., Athanasiou M., Schultz R., Roe B.A., Chen F., Pan H., Ramser J., Lehrach H., Reinhardt R., McCombie W.R., de la Bastide M., Dedhia N., Blöcker H., Hornischer K., Nordsiek G., Agarwala R., Aravind L., Bailey J.A., Bateman A., Batzoglou S., Birney E., Bork P., Brown D.G., Burge C.B., Cerutti L., Chen H.C., Church D., Clamp M., Copley R.R., Doerks T., Eddy S.R., Eichler E.E., Furey T.S., Galagan J., Gilbert J.G., Harmon C., Hayashizaki Y., Haussler D., Hermjakob H., Hokamp K., Jang W., Johnson L.S., Jones T.A., Kasif S., Kaspryzk A., Kennedy S., Kent W.J., Kitts P., Koonin E.V., Korf I., Kulp D., Lancet D., Lowe T.M., McLysaght A., Mikkelsen T., Moran J.V., Mulder N., Pollara V.J., Ponting C.P., Schuler G., Schultz J., Slater G., Smit A.F., Stupka E., Szustakowski J., Thierry-Mieg D., Thierry-Mieg J., Wagner L., Wallis J., Wheeler R., Williams A., Wolf Y.I., Wolfe K.H., Yang S.P., Yeh R.F., Collins F., Guyer M.S., Peterson J., Felsenfeld A., Wetterstrand K.A., Patrinos A., Morgan M.J., de Jong P., Catanese J.J., Osoegawa K., Shizuya H., Choi S., Chen Y.J.; International Human Genome Sequencing Consortium: Initial sequencing and analysis of the human genome. Nature, 2001; 409: 860-921
[PubMed] [Full Text HTML] [Full Text PDF]
[42] Lee Y., Ahn C., Han J., Choi H., Kim J., Yim J., Lee J., Provost P., Radmark O., Kim S., Kim V.N.: The nuclear RNase III Drosha initiates microRNA processing. Nature, 2003; 425: 415-419
[PubMed]
[43] Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., Le Paslier D., Frezal J., Cohen D., Weissenbach J., Munnich A., Melki J.: Identification and characterization of a spinal muscular atrophy-determining gene. Cell, 1995; 80: 155-165
[PubMed] [Full Text HTML] [Full Text PDF]
[44] Lewis B.P., Green R.E., Brenner S.E.: Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl. Acad. Sci. USA, 2003; 100: 189-192
[PubMed] [Full Text HTML] [Full Text PDF]
[45] Liu J., Carmell M.A., Rivas F.V., Marsden C.G., Thomson J.M., Song J.J., Hammond S.M., Joshua-Tor L., Hannon G.J.: Argonaute2 is the catalytic engine of mammalian RNAi. Science, 2004; 305: 1437-1441
[PubMed]
[46] Lopez-Bigas N., Audit B., Ouzounis C., Parra G., Guigo R.: Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett., 2005; 579: 1900-1903
[PubMed]
[47] Lopez-Fraga M., Martinez T., Jimenez A.: RNA interference technologies and therapeutics: from basic research to products. BioDrugs, 2009; 23: 305-332
[PubMed]
[48] Luco R.F., Allo M., Schor I.E., Kornblihtt A.R., Misteli T.: Epigenetics in alternative pre-mRNA splicing. Cell, 2011; 144: 16-26
[PubMed] [Full Text HTML] [Full Text PDF]
[49] Lutz C.S.: Alternative polyadenylation: a twist on mRNA 3′ end formation. ACS Chem. Biol., 2008; 3: 609-617
[PubMed]
[50] Lynch C.J., Shah Z.H., Allison S.J., Ahmed S.U., Ford J., Warnock L.J., Li H., Serrano M., Milner J.: SIRT1 undergoes alternative splicing in a novel auto-regulatory loop with p53. PLoS One, 2010; 5: e13502
[PubMed] [Full Text HTML] [Full Text PDF]
[51] Mahajna J., Shi B., Bruskin A.: A four-amino-acid insertion in the ligand-binding domain inactivates hRXRbeta and renders dominant negative activity. DNA Cell Biol., 1997; 16: 463-476
[PubMed]
[52] McCracken S., Fong N., Rosonina E., Yankulov K., Brothers G., Siderovski D., Hessel A., Foster S., Shuman S., Bentley D.L.: 5′-capping enzymes are targetted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 1997; 11: 3306-3318
[PubMed] [Full Text HTML] [Full Text PDF]
[53] Meister G., Tuschl T.: Mechanisms of gene silencing by double-stranded RNA. Nature, 2004; 431: 343-349
[PubMed]
[54] Michels S., Schmidt-Erfurth U., Rosenfeld P.J.: Promising new treatments for neovascular age-related macular degeneration. Expert Opin. Investig. Drugs, 2006; 15: 779-793
[PubMed]
[55] Miller V.M., Gouvion C.M., Davison B.L., Paulson H.L.: Targeting Alzheimer’s disease genes with RNA interference: an efficient strategy for silencing mutant alleles. Nucleic Acids Res., 2004; 32: 661-668
[PubMed] [Full Text HTML] [Full Text PDF]
[56] Misteli T., Spector D.L.: RNA polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol. Cell., 1999; 3: 697-705
[PubMed] [Full Text HTML] [Full Text PDF]
[57] Moller L.B., Tumer Z., Lund C., Petersen C., Cole T., Hanusch R., Seidel J., Jensen L.R., Horn N.: Similar splice-site mutations of the ATP7A gene lead to different phenotypes: classical Menkes disease or occipital horn syndrome. Am. J. Hum. Genet., 2000; 66: 1211-1220
[PubMed] [Full Text HTML] [Full Text PDF]
[58] Munoz M.J., de la Mata M., Kornblihtt A.R.: The carboxy terminal domain of RNA polymerase II and alternative splicing. Trends Biochem. Sci., 2010; 35: 497-504
[PubMed]
[59] Narla G., Difeo A., Reeves H.L., Schaid D.J., Hirshfeld J., Hod E., Katz A., Isaacs W.B., Hebbring S., Komiya A., McDonnell S.K., Wiley K.E., Jacobsen S.J., Isaacs S.D., Walsh P.C., Zheng S.L., Chang B.L., Friedrichsen D.M., Stanford J.L., Ostrander E.A., Chinnaiyan A.M., Rubin M.A., Xu J., Thibodeau S.N., Friedman S.L., Martignetti J.A.: A germline DNA polymorphism enhances alternative splicing of the KLF6 tumor suppresor gene and is associated with increased prostate cancer risk. Cancer Res., 2005; 65: 1213-1222
[PubMed] [Full Text HTML] [Full Text PDF]
[60] Narla G., DiFeo A., Yao S., Banno A., Hod E., Reeves H.L., Qiao R.F., Camacho-Vanegas O., Levine A., Kirschenbaum A., Chan A.M., Friedman S.L., Martignetti J.A.: Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res., 2005; 65: 5761-5768
[PubMed] [Full Text HTML] [Full Text PDF]
[61] Navaratnam D.S., Bell T.J., Tu T.D., Cohen E.L., Oberholtzer J.C.: Differential distribution of Ca2+-activated K+ channel splice variants among hair cells along the tonopic axis of the chick cochlea. Neuron, 1997; 19: 1077-1085
[PubMed] [Full Text HTML] [Full Text PDF]
[62] Nicholls C.D., Shields M.A., Lee P.W., Robbins S.M., Beattie T.L.: UV-dependent alternative splicing uncoples p53 activity and PIG3 gene function through rapid proteolytic degradation. J. Biol. Chem., 2004; 279: 24171-24178
[PubMed] [Full Text HTML] [Full Text PDF]
[63] Nilsen T.W.: The spliceosome: the most complex macromolecular machine in the cell? Bioessays, 2003; 25: 1147-1149
[PubMed]
[64] Nilsen T.W., Graveley B.R.: Expansion of the eukariotic proteome by alternative splicing. Nature, 2010; 463: 457-463
[PubMed]
[65] Nissim-Rafinia M., Kerem B.: Splicing regulation as a potential genetic modifier. Trends. Genet., 2002; 18: 123-127
[PubMed]
[66] Nowak K.J., Davies K.E.: Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep., 2004; 5: 872-876
[PubMed] [Full Text HTML] [Full Text PDF]
[67] Okazaki Y., Furuno M., Kasukawa T., Adachi J., Bono H., Kondo S., Nikaido I., Osato N., Saito R., Suzuki H., Yamanaka I., Kiyosawa H., Yagi K., Tomaru Y., Hasegawa Y., Nogami A., Schönbach C., Gojobori T., Baldarelli R., Hill D.P., Bult C., Hume D.A., Quackenbush J., Schriml L.M., Kanapin A., Matsuda H., Batalov S., Beisel K.W., Blake J.A., Bradt D., Brusic V., Chothia C., Corbani L.E., Cousins S., Dalla E., Dragani T.A., Fletcher C.F., Forrest A., Frazer K.S., Gaasterland T., Gariboldi M., Gissi C., Godzik A., Gough J., Grimmond S., Gustincich S., Hirokawa N., Jackson I.J., Jarvis E.D., Kanai A., Kawaji H., Kawasawa Y., Kedzierski R.M., King B.L., Konagaya A., Kurochkin I.V., Lee Y., Lenhard B., Lyons P.A., Maglott D.R., Maltais L., Marchionni L., McKenzie L., Miki H., Nagashima T., Numata K., Okido T., Pavan W.J., Pertea G., Pesole G., Petrovsky N., Pillai R., Pontius J.U., Qi D., Ramachandran S., Ravasi T., Reed J.C., Reed D.J., Reid J., Ring B.Z., Ringwald M., Sandelin A., Schneider C., Semple C.A., Setou M., Shimada K., Sultana R., Takenaka Y., Taylor M.S., Teasdale R.D., Tomita M., Verardo R., Wagner L., Wahlestedt C., Wang Y., Watanabe Y., Wells C., Wilming L.G., Wynshaw-Boris A., Yanagisawa M., Yang I., Yang L., Yuan Z., Zavolan M., Zhu Y., Zimmer A., Carninci P., Hayatsu N., Hirozane-Kishikawa T., Konno H., Nakamura M., Sakazume N., Sato K., Shiraki T., Waki K., Kawai J., Aizawa K., Arakawa T., Fukuda S., Hara A., Hashizume W., Imotani K., Ishii Y., Itoh M., Kagawa I., Miyazaki A., Sakai K., Sasaki D., Shibata K., Shinagawa A., Yasunishi A., Yoshino M., Waterston R., Lander E.S., Rogers J., Birney E., Hayashizaki Y., FANTOM Consortium, RIKEN Genome Exploration Research Group Phase I & II Team: Analysis of the mouse transctiptome based on functional annotation of 60,770 full-lenght cDNAs. Nature, 2002; 420: 563-573
[PubMed] [Full Text HTML] [Full Text PDF]
[68] Olivares-Illana V., Fahraeus R.: p53 isoforms gain functions. Oncogene, 2010; 29: 5113-5119
[PubMed]
[69] Pagani F., Raponi M., Baralle F.E.: Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. Proc. Natl. Acad. Sci. USA, 2005; 102: 6368-6372
[PubMed] [Full Text HTML] [Full Text PDF]
[70] Pandaya-Jones A., Black D.L.: Co-transciptional splicing of constitutive and alternative exons. RNA, 2009; 15: 1896-1908
[PubMed] [Full Text HTML] [Full Text PDF]
[71] Pandit S., Wang D., Fu X.D.: Functional integration of transcription and RNA processing machineries. Curr. Opin. Cell Biol., 2008; 20: 260-265
[PubMed] [Full Text HTML] [Full Text PDF]
[72] Park J.W., Graveley B.R.: Complex alternative splicing. Adv. Exp. Med. Biol., 2007; 623: 50-63
[PubMed]
[73] Pasqualini C., Guivarc’h D., Barnier J.V., Guibert B., Vincent J.D., Vernier P.: Differential subcellular distribution and transcriptional activity of sigmaE3, sigmaE4, and sigmaE3-4 isoforms of the rat estrogen receptor-alpha. Mol. Endocrinol., 2001; 15: 894-908
[PubMed] [Full Text HTML] [Full Text PDF]
[74] Prou D., Gu W.J., Le Crom S., Vincent J.D., Salamero J., Vernier P.: Intracellular retention of the two isoforms of the D(2) dopamine receptor promotes endoplasmic reticulum disruption. J. Cell Sci., 2001; 114: 3517-3527
[PubMed] [Full Text HTML] [Full Text PDF]
[75] Proudfoot N.: New perspectives on connecting messenger RNA 3′ end formation to transcription. Curr. Opin. Cell Biol., 2004; 16: 272-278
[PubMed]
[76] Raharjo S.B., Emoto N., Ikeda K., Sato R., Yokoyama M., Matsuo M.: Alternative splicing regulates the endoplasmic reticulum localization or secretion of soluble secreted endopeptidase. J. Biol. Chem., 2001; 276: 25612-25620
[PubMed] [Full Text HTML] [Full Text PDF]
[77] Resch A., Xing Y., Alekseyenko A., Modrek B., Lee C.: Evidence for a subpopulation of conserved alternative splicing events under selection pressure for protein reading frame preservation. Nucleic Acids Res., 2004; 32: 1261-1269
[PubMed] [Full Text HTML] [Full Text PDF]
[78] Ritchie D.B., Schellenberg M.J., MacMillan A.M.: Spliceosome structure: piece by piece. Biochim. Biophys. Acta, 2009; 1789: 624-633
[PubMed]
[79] Roberts G.C., Smith C.W.: Alternative splicing: combinatorial output from the genome. Curr. Opin. Chem. Biol., 2002; 6: 375-383
[PubMed]
[80] Rodrigues N.R., Owen N., Talbot K., Ignatius J., Dubowitz V., Davies K.E.: Deletions in the survival motor neuron gene on 5q13 in autosomal recessive spinal muscular atrophy. Hum. Mol. Genet., 1995; 4: 631-634
[PubMed]
[81] Rodriguez-Lebron E., Paulson H.L.: Allele-specific RNA interference for neurological disease. Gene Ther., 2006; 13: 576-581
[PubMed]
[82] Rosenblatt K.P., Sun Z.P., Heller S., Hudspeth A.J.: Distribution of Ca2+-activated K+ channel isoforms along the tonotopic gradient of the chicken’s cochlea. Neuron, 1997; 19: 1061-1075
[PubMed] [Full Text HTML] [Full Text PDF]
[83] Sanford J.R., Caceres J.F.: Pre-mRNA splicing: life at the centre of the dogma. J. Cell Sci., 2004; 117: 6261-6263
[PubMed] [Full Text HTML] [Full Text PDF]
[84] Schellenberg M.J., Ritchie D.B., MacMillan A.M.: Pre-mRNA splicing: a complex picture in higher definition. Trends Biochem. Sci., 2008; 33: 243-246
[PubMed]
[85] Schmucker D., Chen B.: Dscam and DSCAM: complex genes in simple animals, complex animals yet simple genes. Genes Dev., 2009; 23: 147-156
[PubMed] [Full Text HTML] [Full Text PDF]
[86] Scholz H., Kirschner K.M.: Oxygen-dependent gene expression in development and cancer: lessons learned from the Wilm’s tumor gene, WT1. Front. Mol. Neurosci., 2011; 4: 4
[PubMed] [Full Text HTML] [Full Text PDF]
[87] Sharp P.A.: The discovery of split genes and RNA splicing. Trends Biochem. Sci., 2005; 30: 279-281
[PubMed]
[88] Singh A., Karnoub A.E., Palmby T.R., Lengyel E., Sondek J., Der C.J.: Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene, 2004; 23: 9369-9380
[PubMed]
[89] Slamon D.J., Godolphin W., Jones L.A., Holt J.A., Wong S.G., Keith D.E., Levin W.J., Stuart S.G., Udove J., Ullrich A., Press M.F.: Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science, 1989; 244: 707-712
[PubMed]
[90] Smoliński D.J., Wróbel B., Zienkiewicz K., Niedojadlo J.: Organizacja systemu splicingowego w komórkach linii generatywnej. Kosmos, 2003; 52: 481-492
[91] Sperling J., Azubel M., Sperling R.: Structure and function of the pre-mRNA splicing machine. Structure, 2008; 16: 1605-1615
[PubMed] [Full Text HTML] [Full Text PDF]
[92] Srebrow A., Kornblihtt A.R.: The connection between splicing and cancer. J. Cell Sci., 119: 2635-2641
[PubMed] [Full Text HTML] [Full Text PDF]
[93] Stamm S., Ben-Ari S., Rafalska I., Tang Y., Zhang Z., Toiber D., Thanaraj T.A., Soreq H.: Function of alternative splicing. Gene, 2005; 344: 1-20
[PubMed]
[94] Stanek D., Neugebauer K.M.: The Cajal body: a meeting place for spliceosomal snRNPs in the nuclear maze. Chromosoma, 2006; 115: 343-354
[PubMed]
[95] Stetefeld J., Ruegg M.A.: Structural and functional diversity generated by alternative mRNA splicing. Trends Biochem. Sci., 2005; 30: 515-521
[PubMed]
[96] Sureau A., Gattoni R., Dooghe Y., Stevenin J., Soret J.: SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J., 2001; 20: 1785-1796
[PubMed] [Full Text HTML] [Full Text PDF]
[97] Taliaferro J.M., Alvarez N., Green R.E., Blanchette M., Rio D.C.: Evolution of a tissue-specific splicing network. Genes Dev., 2011; 25: 608-620
[PubMed] [Full Text HTML] [Full Text PDF]
[98] Tazi J., Bakkour N., Stamm S.: Alternative splicing and disease. Biochim. Biophys. Acta, 2009; 1792: 14-26
[PubMed]
[99] Tijsterman M., Plasterk R.H.: Dicers at RISC; the mechanism of RNAi. Cell, 2004; 117: 1-3
[PubMed] [Full Text HTML] [Full Text PDF]
[100] Toor N., Keating K.S., Pyle A.M.: Structural insights into RNA splicing. Curr. Opin. Struct. Biol., 2009; 19: 260-266
[PubMed] [Full Text HTML] [Full Text PDF]
[101] Valadkhan S.: snRNAs as the catalysts of pre-mRNA splicing. Curr. Opin. Chem. Biol., 2005; 9: 603-608
[PubMed]
[102] Wachtel C., Manley J.L.: Splicing of mRNA precursors: the role of RNAs and proteins in catalysis. Mol. Biosyst., 2009; 5: 311-316
[PubMed]
[103] Wakiyama M., Imataka H., Sonenberg N.: Interaction of eIF4G with poly(A)-binding protein stimulates translation and is critical for Xenopus oocyte maturation. Curr. Biol., 2000; 10: 1147-1150
[PubMed] [Full Text HTML] [Full Text PDF]
[104] Wang G.S., Cooper T.A.: Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet., 2007; 8: 749-761
[PubMed]
[105] Wang Z., Burge C.B.: Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA, 2008; 14: 802-813
[PubMed] [Full Text HTML] [Full Text PDF]
[106] Ward A.J., Cooper T.A.: The pathobiology of splicing. J. Pathol., 2010; 220: 152-163
[PubMed] [Full Text HTML] [Full Text PDF]
[107] Wood C.M., Goodman P.A., Vassilev A.O., Uckun F.M.: CD95 (APO-1/FAS) deficiency in infant acute lymphoblastic leukemia: detection of novel soluble Fas splice variants. Eur. J. Haematol., 2003; 70: 156-171
[PubMed]
[108] Wood M., Yin H., McClorey G.: Modulating the expression of disease genes with RNA-based therapy. PLoS Genet., 2007; 3: e109
[PubMed] [Full Text HTML] [Full Text PDF]
[109] Xu Q., Modrek B., Lee C.: Genome-wide detection of tissue-specific alternative splicing in the human transctiptome. Nucleic Acids Res., 2002; 30: 3754-3766
[PubMed] [Full Text HTML] [Full Text PDF]
[110] Zhang X.H., Chasin L.A.: Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev., 2004; 18: 1241-1250
[PubMed] [Full Text HTML] [Full Text PDF]
[111] Zhu H., Guo W., Zhang J.J., Davis J.J., Teraishi F., Wu S., Cao X., Daniel J., Smythe W.R., Fang B.: Bcl-XL small interfering RNA suppresses the proliferation of 5-fluorouracil-resistant human colon cancer cells. Mol. Cancer Ther., 2005; 4: 451-456
[PubMed]
[112] Zhu H., Guo W., Zhang L., Davis J.J., Wu S., Teraishi F., Cao X., Smythe W.R., Fang B.: Enhancing TRAIL-induced apoptosis by Bcl-X(L) siRNA. Cancer Biol. Ther., 2005; 4: 393-397
[PubMed] [Full Text PDF]
[113] Zienkiewicz K., Niedojadło J.: Natura i funkcje ciał Cajala w świetle nowych badań. Post. Biol. Kom., 2004; 31: 313-327
[Abstract]
Autorzy deklarują brak potencjalnych konfliktów interesów.