
Rola białek IRP (iron regulatory proteins) w regulacji ogólnoustrojowej homeostazy żelaza: lekcje płynące z badań na myszach z nokautem genów Irp1 i Irp2
Paweł Lipiński 1 , Rafał Starzyński 1Streszczenie
W komórkach ssaków metabolizm żelaza jest regulowany przez dwa cytoplazmatyczne białka IRP1 i IRP2 (iron regulatory proteins). Białka te wiążą się do swoistych sekwencji RNA, określanych jako sekwencje reagujące na żelazo (iron responsive element – IREs), występujących w niepodlegających translacji regionach (UTR) 3’ i 5’ kilku mRNA, m.in. mRNA kodujących ferrytynę i receptor transferyny. Przy niedoborze żelaza w komórce białka IRP wiążą się z IRE występującymi w 5’ UTR mRNA ferrytyny i blokują translację tego mRNA oraz z sekwencjami IRE w 3’UTR receptora transferyny, co powoduje stabilizację tego mRNA. Odwrotna regulacja zarówno ferrytyny, jak i receptora transferyny, która jest wynikiem braku interakcji białek IRP z sekwencjami IRE odbywa się w komórkach o dużym stężeniu żelaza. Następstwem regulacji za pośrednictwem białek IRP, zarówno w warunkach niedoboru jak i nadmiaru żelaza w komórce, jest szybki powrót komórkowego poziomu żelaza do normy. Wiedza o roli białek IRP w utrzymaniu komórkowej równowagi pochodzi głównie z badań na komórkach hodowanych in vitro. Znaczenie białek IRP w regulacji ogólnoustrojowego metabolizmu żelaza, a w szczególności w sygnalizacji między komórkami o kluczowym znaczeniu dla obiegu żelaza w organizmie, takimi jak enterocyty dwunastnicy, makrofagi układu siateczkowo-śródbłonkowego, hepatocyty i prekursory erytrocytów w szpiku kostnym dopiero zaczyna być przedmiotem badań. W pracy przedstawiono charakterystykę metabolizmu żelaza u myszy z nokautem genów Irp1 i Irp2. Podkreślono znaczenie tych mysich modeli doświadczalnych w poznaniu roli białek IRP w fizjologii żelaza u ssaków
Słowa kluczowe:ferrytyna • IRE • IRP • metabolizm żelaza • myszy • neurodegeneracja • niedokrwistość • nokaut • receptor transferyny
Summary
The iron regulatory proteins (IRP1 and IRP2) are two cytoplasmic RNA-binding proteins that control iron metabolism in mammalian cells. Both IRPs bind to specific sequences, called ironresponsive elements (IREs), located in the 3’ or 5’ untranslated regions (UTR) of several mRNAs, in particular the mRNA encoding ferritin subunits and transferrin receptor. At low intracellular iron concentration, IRPs bind to the IRE of ferritin mRNA at its 5’-UTR and block translation, whereas they stabilize transferrin receptor mRNA through direct interactions with several IRE motifs in the 3’-UTR. The converse regulation of ferritin and TfR synthesis, resulting from lack of binding of IRPs to IRE, occurs in cells with high iron level. In both, iron deficiency and excess IRP-mediated regulation rapidly restore the physiological cytosolic iron level. The role of IRPs in maintaining the intracelluar iron balance has been well characterized in numerous types of mammalian cells in culture. However, the importance of IRPs in the regulation of systemic iron metabolism in mammals, in particular in signaling between cells which play major roles in body iron metabolism, such as duodenal enterocytes, reticuloendothelial macrophages, hepatocytes, and bone marrow precursors of red blood cells, is only beginning to be investigated. This review presents the basic features of iron metabolism in IRP1 and IRP2 knockout mice and focuses on how recent studies on these animal models have advanced our understanding of the role of IRPs in iron mammalian physiology.
Key words:anemia • ferritin • IRE • iron metabolism • IRP • knock out • mice • neurodegeneration • transferrin receptor
WSTĘP
Żelazo jest elementem nieorganicznym, niezbędnym do przebiegu wielu reakcji biochemicznych w zdecydowanej większości organizmów żywych. Jedynymi znanymi organizmami niewymagającymi żelaza są bakterie z rodzaju Lactobacillus [62]. O wykorzystaniu żelaza przez organizmy żywe decyduje niezwykle duża zmienność potencjału oksydoredukcyjnego (redoks) układu dwóch głównych postaci żelaza jonowego: Fe2+(jonu żelazawego)/Fe3+(jonu żelazowego). Potencjał redoks pary Fe2+/Fe3+ może być precyzyjnie dostosowany do specyfiki konkretnej reakcji przez ligandy wiążące żelazo oraz przez ładunek elektryczny i strukturę przestrzenną aminokwasów znajdujących się w bezpośrednim otoczeniu. Białkowe centra aktywne, w których występuje żelazo obejmują niemal cały zakres potencjałów redoks występujących w układach biologicznych – od około –0,5 do około +0,6 V [46]. Do białek, których funkcję i aktywność wyznaczają wbudowane w ich cząsteczki atomy żelaza należą m.in. białka transportujące tlen cząsteczkowy (O2) (hemoglobina i mioglobina), enzymy mitochondrialnego łańcucha oddechowego (cytochromy), enzymy cyklu Krebsa (akonitaza mitochondrialna), enzymy inaktywujące toksyczne postaci O2 (katalaza), enzymy biorące udział w syntezie DNA (reduktaza rybonukleotydowa). Niezbędność tych wszystkich białek do prawidłowego funkcjonowania organizmów żywych jest odzwierciedleniem biologicznego znaczenia żelaza.
Czynnikiem, który określił kształt metabolizmu żelaza u współcześnie żyjących organizmów żywych jest O2. O ile jon Fe2+ jest bardzo dobrze rozpuszczalny w wodzie, o tyle jon Fe3+, dominująca postać jonowego żelaza w środowisku tlenowym jest nierozpuszczalny w roztworach wodnych o fizjologicznym pH. Kolejnym niekorzystnym elementem współistnienia tlenu i żelaza w układach biologicznych jest generowanie rodnika hydroksylowego (˙OH) w obecności jonów Fe2+ i częściowo zredukowanych form O2, takich jak anionorodnik ponadtlenkowy (O2˙–) i nadtlenek wodoru (H2O2). Prawdopodobny mechanizm powstawania rodnika ˙OH in vivo opisują dwie reakcje określane często jako cykl Fentona [4], w nawiązaniu do obserwacji poczynionych przez niemieckiego chemika Fentona, który pod koniec XIX w. opisał silne właściwości utleniające mieszaniny soli żelazawych i H2O2 [14]. Pierwsza z tych reakcji to właściwa reakcja Fentona, druga opisuje mechanizm regeneracji jonu Fe2+ przez O2˙–.

Rodnik ˙OH jest jedną z najbardziej reaktywnych chemicznie cząsteczek istniejących w przyrodzie. Charakteryzuje się wysokim potencjałem redoks, co wskazuje na jego bardzo silne właściwości utleniające i na możliwość reakcji z większością związków organicznych. Reakcje te charakteryzuje bardzo mała swoistość. Szybkość tych reakcji jest kontrolowana tylko przez szybkość dyfuzji rodnika ˙OH. W tym kontekście jego zmiataczem stanie się każda przypadkowa cząsteczka, która się znajdzie w bezpośrednim miejscu jego powstania.
POTRANSKRYPCYJNY MECHANIZM REGULACJI KOMÓRKOWEGO METABOLIZMU ŻELAZA
Biologiczna niezbędność żelaza, a także jego potencjalna toksyczność wymagają ścisłej kontroli nad metabolizmem tego biometalu. Główny komórkowy mechanizm tej kontroli funkcjonuje w oparciu o 2 białka regulatorowe (iron regulatory proteins – IRPs) – IRP1 i IRP2, które opisano pod koniec lat 80. ub.w. Charakterystyka IRPs oraz ich rola w potranskrypcyjnej regulacji syntezy białek związanych z metabolizmem żelaza są przedmiotem licznych artykułów przeglądowych [7,9,13], w tym kilku w piśmiennictwie polskojęzycznym [34,35,58]. Z tego powodu ograniczymy się jedynie do zwięzłego omówienia tej regulacji, odsyłając czytelników zainteresowanych szczegółami do wyżej cytowanych prac przeglądowych.
Molekularny mechanizm kontroli ekspresji genów kodujących białka związane z metabolizmem żelaza jest oparty na jednoczesnej regulacji translacji lub stabilności gotowych transkryptów. Oprócz IRPs, głównymi jego elementami są sekwencje IRE (iron responsive element), których występowanie stwierdzono w niepodlegających translacji regionach (untranslated regions – UTR) 5’ lub 3’ mRNA podjednostek ferrytyny (Ft) [13], receptora transferyny (TfR) [13], ferroportyny [38], DMT1 (jednej z izoform tego mRNA powstałej w wyniku różnego składania pierwotnego transkryptu) [21], syntetazy d
-aminolewulinowej [12], akonitazy mitochondrialnej [63], białka prekursorowego amyloidu [60], podjednostki b dehydrogenazy bursztynianowej [29] i oksydazy glikolowej myszy [30]. Istotą skoordynowanej regulacji syntezy białek przez system IRP/IRE jest hamowanie translacji transkryptu, gdy interakcja między IRP a IRE zachodzi w końcu 5’UTR oraz stabilizacja transkryptu, gdy miejscem interakcji jest koniec 3’UTR. Funkcjonowanie tego potranskrypcyjnego mechanizmu poznano najlepiej w odniesieniu do regulacji syntezy ferrytyny i receptora transferyny
IRP1 jest białkiem dwufunkcyjnym, które występuje w cytoplazmie jako akonitaza, enzym zawierający katalityczne centrum [4Fe-4S] (holo-IRP1) lub jako białko wiążące się z IRE, pozbawione tego centrum (apo-IRP1). Dwie aktywności IRP1 wzajemnie się wykluczają i są odwrotnie regulowane przez jony żelaza zawarte w zmiennej puli żelaza (labile iron pool – LIP). Przy niedoborze jonów żelaza w komórce przeważa apo-IRP1, które przyłącza się do IREs, występujących w mRNA podjednostek ferrytyny oraz receptora transferyny. Gdy IRP1 wiąże się z IRE w końcu 5’UTR mRNA podjednostek Ft powoduje zahamowanie ich translacji i syntezy Ft. Wiązanie IRP1 z IRE występującymi w końcu 3’UTR mRNA TfR zwiększa jego stabilność a w konsekwencji syntezę receptora. Odwrotna regulacja syntezy obu białek wynikająca z braku wiązania holo-IRP1 z IRE, odbywa się, gdy stężenie LIP w komórce jest duże [13,58].
Mimo znacznego podobieństwa IRP2 do IRP1, białko to nie ma centrum żelazowo-siarkowego i nie wykazuje aktywności akonitazy [23]. Zmiany w wiązaniu się IRP2 z IRE, będące następstwem zmian w poziomie LIP nie wynikają z potranslacyjnej konwersji między dwoma postaciami białka, tak jak się dzieje w wypadku IRP1. Regulacja wiązania się IRP2 z RNA przez żelazo odbywa się w następstwie degradacji tego białka w proteasomach [22,27]. W komórkach o wysokim poziomie żelaza degradacja IRP2 ma ten sam skutek biologiczny w odniesieniu do syntezy Ft i TfR, jak w przypadku IRP1 przesunięcie równowagi dwóch postaci tego białka w kierunku holo-IRP1. Przy niedoborze żelaza poziom IRP2 ulega stabilizacji, co ma analogiczny skutek do aktywacji IRP1, i co powoduje zablokowanie syntezy podjednostek Ft i stabilizację mRNA TfR. Zarówno przy niedoborze jak i nadmiarze jonów żelaza skutkiem regulacji syntezy Ft i TfR przez IRP1 i IRP2 jest szybki powrót do fizjologicznego poziomu LIP.
NIEZNANA ROLA SYSTEMU IRP/IRE WE WSPÓŁDZIAŁANIU KOMÓREK W OBIEGU ŻELAZA W ORGANIZMIE
Utrzymanie ogólnoustrojowej homeostazy żelaza opiera się na współdziałaniu komórek różnych typów o ściśle wyspecjalizowanych funkcjach związanych z transportem żelaza z przewodu pokarmowego (enterocyty absorpcyjne), jego magazynowaniem (hepatocyty), reutylizacją (komórki układu siateczkowo-śróbłonkowego śledziony i wątroby) oraz syntezą hemoglobiny (erytroblasty). Enterocyty absorpcyjne dwunastnicy zaopatrujące cały organizm w żelazo, pobierają je z diety, a następnie uwalniają do układu krążenia. Są to komórki spolaryzowane, które na odcinku błony komórkowej kontaktującym się ze światłem jelita oraz na odcinku zwróconym w stronę naczyń krwionośnych mają odrębne tandemy białek importujących żelazo: dwunastniczy cytochrom b (Dcytb), transporter metali dwuwartościowych (DMT1) i uwalniających żelazo: hefajstyna (Heph), ferroportyna (Fpn) [52]. Erytroblasty to komórki zaprogramowane na intensywne pobieranie żelaza związanego z transferyną, które następnie w znacznej części wykorzystują do syntezy hemu. Erytroblasty charakteryzują się szczególnie dużą gęstością TfR na błonach komórkowych [48]. Makrofagi wątroby i śledziony fagocytują stare erytrocyty i odprowadzają do krążenia żelazo uwolnione w wyniku degradacji hemu. Jest to możliwe dzięki dużej aktywności oksygenazy hemowej 1 (HO-1), podstawowego enzymu biorącego udział w tym procesie [37] oraz dzięki znaczącej ekspresji ferroportyny na błonie komórkowej [1]. Są to komórki o niezwykle intensywnym metabolizmie żelaza. W wyniku recyrkulacji żelaza przez makrofagi w organizmie dorosłego człowieka do krwi dostaje się w ciągu doby około 20 mg żelaza, czyli 10-krotnie więcej niż wynosi dobowa absorpcja żelaza przez enterocyty [28]. Wreszcie, hepatocyty to komórki o dużym potencjale gromadzenia żelaza w postaci ferrytyny, które może być uwalniane dla potrzeb całego organizmu w sytuacji niedoboru tego mikroelementu [2,3].
O ile funkcjonowanie systemu IRP/IRE poznano gruntownie w odniesieniu do komórek (wszystkich wymienionych wyżej typów) hodowanych in vitro, o tyle jego rola, a w szczególności udział poszczególnych IRP w koordynowaniu ogólnoustrojowej homeostazy żelaza u ssaków pozostaje nieznana. W komórkach każdego z wyżej wymienionych typów występuje zarówno IRP1 jak i IRP2, chociaż ekspresja każdego z tych białek, a także wzajemny stosunek ekspresji IRP1 do IRP2 są silnie zróżnicowane [26,39,50]. W każdej komórce określona aktywność wiązania się białek IRP z sekwencjami IRE wyznacza intensywność syntezy ferrytyny (a także innych białek, kodowanych przez mRNA zawierające sekwencje IRE), która jest głównym białkiem determinującym fizjologiczny poziom LIP w komórce [45]. Wydaje się, że w wypadku niektórych komórek, takich jak enterocyty absorpcyjne, hepatocyty i makrofagi układu siateczkowo-śródbłonkowego poziom LIP musi uwzględniać nie tylko indywidualne potrzeby tych komórek, ale również ich funkcje wynikające z roli, jaką pełnią w ogólnoustrojowym metabolizmie żelaza.
I tak w enterocytach absorpcyjnych, system IRP/IRE kontroluje wielkość puli wolnego żelaza, które podlega transportowi do krwi. Odbywa się to przez odpowiednią regulację poziomu ferrytyny w tych komórkach, która wiążąc żelazo transportowane do enterocytów z przewodu pokarmowego z udziałem białka DMT1, ogranicza jego ilość podlegającą dalszej absorpcji do krwiobiegu z udziałem ferroportyny. W świetle ostatnich badań hepatocyty są głównymi, choć nie jedynymi komórkami ssaków syntetyzującymi hepcydynę, peptyd kodowany przez gen Hamp, którego ekspresja indukowana jest przez żelazo, co powoduje zwiększenie jego retencji w enterocytach i makrofagach [36,51]. W obu przypadkach molekularny mechanizm działania hepcydyny opiera się na jej interakcji z ferroportyną, jedynym znanym obecnie białkiem transportującym żelazo z komórki do środowiska pozakomórkowego. Oddziaływanie hepcydyny na ferroportynę powoduje przemieszczenie tego białka z błony komórkowej do cytoplazmy i przyspieszoną jego biodegradację [43]. Przypuszczalnie to właśnie LIP stanowi w hepatocytach tę frakcję żelaza komórkowego, która wpływa na ekspresję genu Hamp. Poziom LIP w tych komórkach musi podlegać zatem takiej regulacji przez białka IRP, aby wskutek odpowiedniej ekspresji, sekrecji a następnie oddziaływania hepcydyny na ferroportynę występującą na błonach komórkowych enterocytów i makrofagów, ilość żelaza uwolnionego z tych komórek była zsynchronizowana z intensywnością wykorzystania żelaza w syntezie hemu w erytroblastach. Z kolei w makrofagach układu siateczkowo-śródbłonkowego, funkcjonowanie systemu IRP/IRE ukierunkowane jest na detoksyfikację żelaza (intensywna synteza ferrytyny) uwolnionego wskutek biodegradacji hemu zawartego w sfagocytowanych erytrocytach. Jest to szczególnie istotne w sytuacjach patologicznych, takich jak przewlekłe stany zapalne, w czasie których synteza hepcydyny jest indukowana niezależnie od żelaza przez interleukinę 6 [42]. Dochodzi wówczas do nadmiernej akumulacji żelaza w tych komórkach.
Jedynym znanym jak dotąd przykładem ogólnoustrojowej patologii będącej konsekwencją zaburzenia w funkcjonowaniu systemu IRP/IRE jest występujący u ludzi zespół dziedzicznej katarakty związanej z hiperferrytynemią (hereditary hyperferritinemia cataract syndrome), którego objawy przypominają kataraktę, z towarzyszącym odkładaniem się ferrytyny o przewadze podjednostki L w soczewce gałki ocznej [33]. Molekularnym podłożem tego zaburzenia jest mutacja (substytucja A/G) w sekwencji IRE, a ściślej w konserwatywnej sekwencji 6-nukleotydowej pętli IRE (która jako całość jest strukturą typu szpilki do włosów – hairpin strucure), występującej w końcu 5’UTR mRNA podjednostki L ferrytyny. Mutacja ta uniemożliwia interakcję IRE z IRP1, czego skutkiem jest niekontrolowana, masowa synteza podjednostek L-Ft [5]. Jak dotąd nie stwierdzono dysregulacji sytemu IRP/IRE i w konsekwencji zaburzeń metabolizmu żelaza na poziomie całego organizmu, u podłoża których leżałyby mutacje genów Irp1 i Irp2. Wiedza na temat roli poszczególnych IRP w regulacji ogólnoustrojowego metabolizmu żelaza u ssaków pochodzi zatem z przeprowadzonych w ostatnich latach badań na modelach mysich z nokautami genów kodujących te białka.
HOMEOSTAZA ŻELAZA U MYSZY Z NOKAUTEM GENU IRP1
Charakterystykę ogólnoustrojowego metabolizmu żelaza u myszy z nokautem genu Irp1 jako pierwsi przedstawili Meyron-Holtz i wsp. [39]. Myszy KO IRP1 otrzymano za pośrednictwem homologicznej rekombinacji w embrionalnych komórkach macierzystych (EC), po ich transfekcji wektorem delecyjnym pPNT, zawierającym gen oporności na neomycynę oraz flankujące fragmenty genu Irp1. Tak otrzymany zmutowany gen Irp1 był o 6 kb krótszy od genu dzikiego. Otrzymane chimerowe potomstwo, krzyżowano wstecznie z myszami szczepu C57BL/6. W wyniku krzyżowania heterozygot Irp1+/– otrzymano potomstwo, wśród którego proporcja poszczególnych genotypów była zgodna z rozkładem mendlowskim. Badania histologiczne głównych organów i gruczołów myszy z nokautem genu Irp1 nie wykazywały nieprawidłowości. Barwienie preparatów tkankowych pochodzących od tych myszy błękitem pruskim (barwnik Perlsa) wykazało prawidłową zawartość i dystrybucję żelaza. Wartości podstawowych parametrów hematologicznych nie odbiegały od normy. Podobnie wartości wskaźników biochemicznych osocza krwi nie wykazywały istotnych różnic w porównaniu do myszy dzikich. W cytowanej pracy dokonano analizy ekspresji tkankowej IRP1 u normalnych myszy i potwierdzono wyniki wcześniejszych badań wskazujących na to, że IRP1 ulega najwyższej ekspresji zarówno na poziomie mRNA, jak i białka w nerce i wątrobie [39,50]. Dodatkowo odnotowano wysoki poziom IRP1 w tkance tłuszczowej umiejscowionej na grzbiecie (brown fat) [39]. U myszy KO IRP1 w tkankach, w których konstytutywna ekspresja IRP1 jest zwykle (u myszy dzikich) największa, spodziewano się zaburzenia syntezy białek, kodowanych przez mRNA zawierający sekwencje IRE. Tymczasem obserwowano jedynie dysregulację syntezy ferrytyny, objawiającą się podwyższonym stężeniem podjednostek H i L w nerkach i tkance tłuszczowej myszy KO IRP1, a więc w tych tkankach, w których u normalnych myszy oprócz wysokiej ekspresji IRP1, ilościowa proporcja IRP1 do IRP2 jest również wysoka. Co ciekawe, w wątrobie – organie o ważnym znaczeniu w utrzymania ogólnoustrojowej homeostazy żelaza [28], nie zaobserwowano zmian w poziomie ferrytyny spowodowanych brakiem IRP1. Niezwykle istotną obserwacją o roli IRP1 w kontroli ogólnoustrojowego metabolizmu żelaza było wykazanie, że u myszy KO IRP1, poddanych diecie pozbawionej żelaza, regulacja syntezy ferrytyny w tkankach odbywa się zgodnie z kanonem regulacyjnym systemu IRP/IRE, co oznacza, że poziom podjednostek ferrytyny był mniejszy, wyłącznie jednak w następstwie zmian aktywności (poziomu) IRP2.
Galy i wsp. [17] przyjęli inną niż Meyron-Holtz i wsp. [39] molekularną strategię unieczynnienia genu Irp1. Zastosowali ukierunkowaną mutagenezę z użyciem techniki gene trap. Poprzez homologiczną rekombinację w embrionalnych komórkach macierzystych E14, do drugiego intronu genu Irp1 wprowadzono konstrukt zawierający kasetę unieczynniającą produkt transkrypcji genu Irp1 oraz gen oporności na neomycynę i gen beta-galaktozydazy. Tak wprowadzony konstrukt naruszał ramkę odczytu w pobliżu N-końca genu Irp1, powodując, że powstały mRNA nie podlegało translacji. Pozytywne klony komórek E14 wprowadzano do embrionów myszy szczepu C57BL6/J. Tak powstałe chimeryczne potomstwo krzyżowano wstecznie z myszami szczepu C57BL6/J. U myszy z dwoma zmutowanymi allelami genu Irp1 obserwowano drastyczne (90%), ale niecałkowite obniżenie poziomu IRP1 [17]. Charakterystykę metabolizmu żelaza u tych myszy opublikowano w formie doniesienia na First Congress of the International BioIron Society, który odbył się w Pradze w 2005 roku [19]. Nie stwierdzono u tych myszy, podobne jak w opisanym wyżej mysim modelu KO IRP1 [39], naruszenia ogólnoustrojowej homeostazy żelaza.
Badania na myszach KO IRP1 przyczyniły się do zasadniczego zwrotu w poglądach dotyczących regulacji metabolizmu żelaza przez to białko, wzbudziły także krytyczną refleksję w stosunku do wniosków płynących z długoletnich badań na komórkach hodowanych in vitro. Badania te, poza nielicznymi wyjątkami [55], wskazywały na IRP1 jako na pierwszoplanowe białko kontrolujące wewnątrzkomórkową homeostazę żelaza w warunkach fizjologicznych oraz przywracające te homeostazę pod wpływem wahań poziomu żelaza. Uwzględniając to, że w komórkach i tkankach IRP1 występuje głównie w postaci akonitazy, a jedynie w 5–15% w postaci apo-IRP1, należy zauważyć, że margines regulacji IRP1 przez nadmiar wolnego żelaza (całkowity zanik postaci apo-IRP1 oraz zwiększenie do 100% postaci enzymatycznej IRP1) jest w istocie niewielki. Z kolei uważna analiza prac, w których udokumentowano indukcje aktywności wiązania się IRP1 z RNA pokazuje, że zjawisko to obserwowano na ogół w komórkach inkubowanych z farmakologicznymi chelatorami żelaza (np. desferal) stosowanymi w stężeniach wielokrotnie przekraczających dawki terapeutyczne.
NEURODEGENERACJA I NIEDOKRWISTOŚĆ U MYSZY Z NOKAUTEM GENU IRP2
IRP2 po raz pierwszy zidentyfikowano w komórkach wątroby szczura [23], a następnie w komórkach ludzkich [54]. Jest to białko o odmiennej, przeważnie mniejszej niż IRP1 ekspresji w tkankach ssaków. Najwyższy poziom IRP2 oznaczono w mózgu [26]. Początkowo IRP2 uważane było – w porównaniu z IRP1 – za białko o drugorzędnym znaczeniu. W ostatnich kilku latach IRP2 skupia jednak na sobie szczególnie duże zainteresowanie badaczy. Jedną z przyczyn stała się zasadnicza zmiana koncepcji, co do molekularnego mechanizmu biodegradacji IRP2 w komórkach ssaków. Jeszcze do niedawna powszechnie akceptowany model degradacji IRP2 uwzględniał katalizowaną przez jony żelaza oksydacyjną modyfikację reszt cysteinowych, w obrębie 73-aminokwasowego fragmentu białka, zwanego domeną degradacyjną. Uważano, że zmodyfikowane IRP2 podlega ubikwitynacji, a następnie degradacji w proteasomach [22]. Nowy model zakłada, że degradacja IRP2 odbywa się z udziałem dioksygenazy 2- oksyglutaranu, enzymu którego aktywność zależy od tlenu. Sygnałem do degradacji IRP2 jest katalizowana przez dioksygenazę hydroksylacja cząsteczki IRP2, umiejscowiona we fragmencie białka nieobejmującym wspomnianej wyżej domeny degradacyjnej [25,61]. Zanim jeszcze zaproponowano nowy mechanizm degradacji IRP2, wykazano, że aktywność tego białka podlega regulacji pod wpływem zmian stężenia tlenu – w warunkach hipoksji wzrasta aktywność IRP2 [24]. Stwierdzono, że u podłoża tej regulacji leży stabilizacja poziomu IRP2, spowodowana zmniejszonym tempem degradacji białka w proteasomach. Niedawne badania wykazały również, że aktywność IRP2 i jego poziom w fibroblastach hodowanych w stężeniu tlenu zbliżonym do atmosferycznego (21%) były znacząco obniżone w porównaniu z fibroblastami mysimi hodowanymi w środowisku o 3–5% zawartości O2, a więc w warunkach zbliżonych do panujących in vivo w tkankach ssaków [40]. Na podstawie tych wyników sformułowano zalecenie do dokonania zasadniczych zmian w metodyce badania aktywności wiązania się IRP2 z IRE, polegających na przygotowywaniu cytoplazmatycznych ekstraktów tkankowych (szczególnie z tych tkanek, w których poziom IRP2 jest niski) w warunkach beztlenowych [6]. Z naszych obserwacji wynika, że rzeczywiście, przestrzeganie tej zasady przyczynia się do uwidocznienia w żelu kompleksów IRP2/IRE, które były niewidoczne, gdy ekstrakty przygotowywano w warunkach atmosferycznego stężenia O2 (ryc. 1). Wydaje się, że nieprzestrzeganie tej zasady w przeszłości mogło być przyczyną wysuwania mylnych wniosków, co do biologicznego znaczenia IRP2 i pomniejszania jego roli w utrzymaniu komórkowej i ogólnoustrojowej homeostazy żelaza.
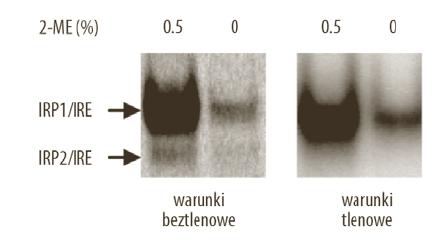
Ryc. 1. Porównanie aktywności wiązania się IRP2 z IRE w ekstraktach cytoplazmatycznych wątroby mysiej przygotowywanych w warunkach tlenowych i beztlenowych. Spontaniczną i całkowitą (indukowaną w obecności 0,5% 2-merkaptoetanolu [8]) aktywność wiązania IRP2 do IRE analizowano w ekstraktach cytoplazmatycznych (5 µg białka) z wątroby mysiej z użyciem techniki opóźnionej migracji sondy RNA (IRE) w żelu [59]. Pobraną wątrobę porcjowano a następnie skrawki natychmiast zamrażano w ciekłym azocie i przechowywano w temp. –80°C. Całą preparatykę ekstraktów z pojedynczych skrawków wątroby przeprowadzano przy atmosferycznym (21%) stężeniu tlenu lub w komorze wypełnionej azotem (N2). Strzałki wskazują kompleksy IRP1 i IRP2 z radioaktywna sondą IRE
Bardziej istotną niż pierwotnie sądzono rolę IRP2 w regulacji metabolizmu żelaza u ssaków sugerują badania na myszach z nokautem genu Irp1. Wynika z nich, że do utrzymania homeostazy żelaza u tych zwierząt wystarcza aktywność trans-regulatorowa IRP2 [39]. U myszy KO IRP1, IRP2 sterują również samodzielnie zmianami w metabolizmie żelaza zachodzącymi pod wpływem zróżnicowanej zawartości tego pierwiastka w diecie [39]. Dalsze argumenty przemawiające za ważną rolą IRP2 w regulacji metabolizmu żelaza przyniosły badania na myszach z nokautem genu Irp2. Jako pierwsi fenotyp myszy Irp2–/– scharakteryzowali La Vaute i wsp. [32]. Myszy uzyskano stosując podobną strategię nokautowania genów, jak w przypadku genu Irp1 [39]. Embrionalne komórki macierzyste (EC), transfekowano wektorem delecyjnym pPNT zawierającym gen oporności na neomycynę oraz flankujące fragmenty genu Irp1. Tak otrzymany zmutowany gen Irp2 był o 11,6 kB krótszy od genu dzikiego Irp2. Otrzymane chimerowe potomstwo, krzyżowano wstecznie ze szczepem myszy C57BL/6. Stwierdzono, że rozwój płodów Irp2–/– przebiega prawidłowo. Ekspresja i aktywność IRP1 w płodowych fibroblastach też jest prawidłowa. Regulacja biosyntezy Ft i TfR odbywa się podobnie jak w fibroblastach płodowych myszy kontrolnych.
Wzrost i rozwój myszy w pierwszych 6 miesiącach życia nie odbiega od normy. Po tym okresie myszy zapadają jednak na postępującą chorobę neurodegeneracyjną. Objawia się ona chwiejnym chodem, wygięciem tylnej części kręgosłupa (kifoza), ataksją, drżeniem, spowolnieniem ruchów, obniżoną siłą uścisku łap. W celu określenia neuropatologicznych podstaw zaburzeń postawy i poruszania się myszy z nokautem genu Irp2 przeprowadzono analizę rozmieszczenia i zawartości żelaza w poszczególnych obszarach i strukturach mózgowia. Chociaż nie stwierdzono zmian w całkowitej zawartości żelaza w tkance mózgowej, to jednak w poszczególnych obszarach istoty białej i szarej wykazano nadmierną akumulację tego metalu, co przedstawia tab. 1.
Tabela 1. Zmiany zawartości żelaza w tkankach myszy KO IRP2 w porównaniu z myszami kontrolnymi
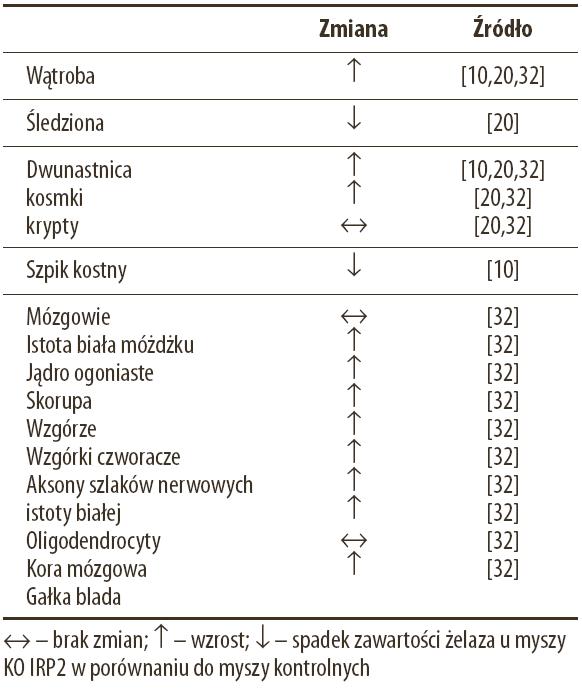
Nagromadzeniu żelaza w większości szlaków nerwowych istoty białej towarzyszyła degeneracja wypustek komórek nerwowowych (aksonów). W istocie białej móżdżku żelazo odkłada się w aksonach oraz w oligodendrocytach (glej skąpokomórkowy) leżących między pęczkami aksonów. Tam też stwierdzono zmiany degeneracyjne. Analiza młodych myszy z nokautem genu Irp2 wykazała, że akumulacja żelaza w aksonach zwykle wyprzedza procesy neurodegenarcyjne, które odnotowano u myszy starszych. Odkładanie się żelaza stwierdzono również w wielu obszarach istoty szarej mózgu (tabela 1). W większości neuronów z tych obszarów obserwowano uszkodzenia błony jądrowej, kurczenie się jąder komórkowych i pojawienie się licznych wakuoli w cytoplazmie. Zwiększona zawartość żelaza w poszczególnych obszarach mózgu pokrywa się ze zwiększoną ekspresją ferrytyny, co stwierdzono metodami immunohistochemicznymi. Prawidłowość tę udokumentowano np. w komórkach Purkiniego móżdżku. W aksonach tych komórek stwierdzono akumulację żelaza i jednocześnie zwiększoną ekspresję ferrytyny. Ponadto w lizatach móżdżku wykazano, metodą Western-blot, podwyższone stężenie ferrytyny i obniżony poziom receptora transferyny. Podsumowując wyniki swoich badań La Vaute i wsp. [32] sugerują, że bezpośrednią przyczyną degeneracji komórek nerwowych u myszy KO IRP2 może być tzw. funkcjonalny niedobór żelaza. Oznacza to, że żelazo w tych komórkach jest związane głównie z ferrytyną, której niekontrolowana synteza, będąca konsekwencją braku głównego represora tego procesu, jakim w mózgu jest IRP2, prowadzi do drastycznego ograniczenia biologicznej dostępności żelaza i zakłócenia przebiegu reakcji biochemicznych opartych na oksydoredukcyjnych właściwościach tego metalu.
Badania Galy’ego i wsp. [20] prowadzone na innym modelu myszy KO IRP2, zarówno młodych (8–10 tygodni) jak i 8-miesięcznych nie potwierdziły obserwacji dotyczących zmian neurodegeneracyjnych obserwowanych przez La Vaute’a i wsp. [32]. Natomiast niewątpliwie zbieżnym elementem badań na dwóch różnych modelach myszy KO IRP2 jest udokumentowanie u tych zwierząt umiarkowanej niedokrwistości mikrocytowej [10,20]. Na występowanie tego zaburzenia wskazują: obniżona wartość hematokrytu, obniżone stężenie hemoglobiny oraz mniejsza objętość erytrocytów (tabela 2).
Tabela 2. Zmiany wartości parametrów hematologicznych u myszy KO IRP2 w porównaniu z myszami kontrolnymi
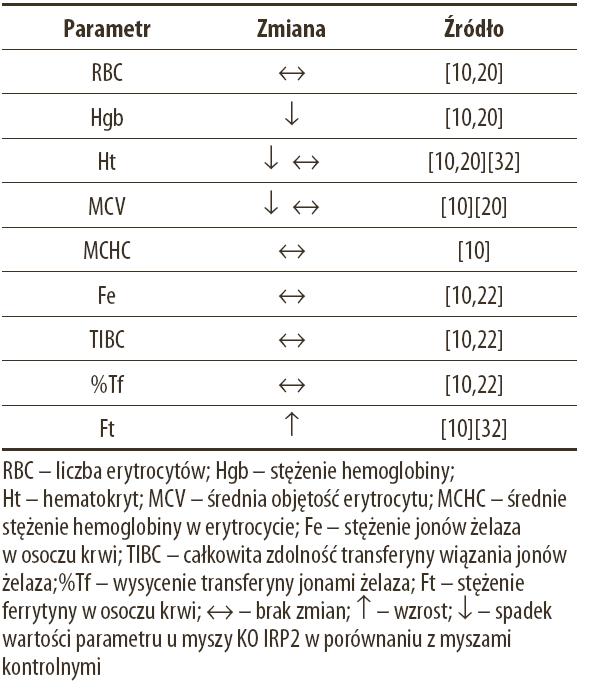
Autorzy zgodnie podkreślają, że niedokrwistość nie jest skutkiem ograniczenia ogólnoustrojowej dostępności żelaza, o czym świadczy nieodbiegąjące od normy stężenie żelaza w osoczu krwi, jak również fizjologiczne wysycenie transferyny jonami żelaza. Za prawdopodobną przyczynę niedokrwistości autorzy uważają naruszenie równowagi żelaza w erytroblastach szpiku kostnego. W komórkach tych rozregulowanie metabolizmu żelaza, będące skutkiem braku IRP2, objawia się podwyższonym stężeniem dwóch podjednostek ferrytyny i obniżonym poziomem receptora transferyny [10,20], co jest bezpośrednią przyczyną niedoboru żelaza niezbędnego do syntezy hemu. Inną konsekwencją braku aktywności IRP2 w komórkach erytroidalnych jest wzrost poziomu syntetazy kwasu aminolewulinowego (ALAS2) [10], enzymu kodowanego przez mRNA zawierający sekwencję IRE w 5’UTR [12] (nieobserwowany przez Galy’ego i wsp. [20]; przyczyną tej rozbieżności może być to, że Cooperman i wsp. [10] analizowali poziom ALAS2 wybiórczo w komórkach erytroidalnych szpiku, a Galy i wsp. [20] w mieszaninie komórek szpiku). ALAS2 jest enzymem inicjującym syntezę hemu poprzez reakcję kondensacji glicyny z bursztynylo-koenzymem A [53]. Zarówno niedobór żelaza jak i nadmierna aktywność ALAS2 prowadzą do zwiększenia poziomu wolnej protoporfiryny IX oraz protoporfiryny zawierającej cynk (który pod nieobecność żelaza jest włączany z udziałem ferrochelatazy do cząsteczek protoporfiryny) [31]. Nadmiar protoporfiryny jest czynnikiem światłouczulającym, który wywołuje porfirie – grupę chorób, objawiających się nadwrażliwością na światło (promienie UVA). U chorych na porfirię obserwuje się w miejscach odsłoniętych wykwity rumieniowe, zwiększoną urażalność skóry i zmiany pęcherzowe, ustępujące często z pozostawieniem blizn [44]. W tym kontekście należy podkreślić, że u myszy KO IRP2 systematycznie obserwowano liczne uszkodzenia oczu i skóry [32]. Autorzy zauważają, że zmiany te mogą stanowić objaw porfirii erytropoetycznej, niezależnej od zaburzeń aktywności ferrochelatazy (enzymu), co zwykle uważa się za przyczynę tej choroby [11].
Biorąc pod uwagę, że absorpcja żelaza z przewodu pokarmowego odbywa się głównie w dwunastnicy, jej funkcjonowanie stanowi niezwykle istotny element w utrzymaniu ogólnoustrojowej homeostazy żelaza. Zaburzenia ekspresji białek biorących udział w transporcie żelaza może prowadzić zarówno do niedokrwistości na tle niedoboru żelaza [16], jak i do uszkodzenia organów wewnętrznych wskutek toksyczności nadmiernie absorbowanego żelaza [47]. W dwunastnicy myszy KO IRP2 wykazano wzrost zawartości żelaza niehemowego w porównaniu z myszami kontrolnymi [20,32]. Barwienie preparatów histologicznych tego fragmentu jelita cienkiego błękitem pruskim, wykazało selektywne występowanie złogów żelaza w enterocytach absorpcyjnych znajdujących się na szczycie kosmków dwunastnicy. Stosując tę samą technikę nie wykryto żelaza w enterocytach występujących na granicy krypt i kosmków [20,32]. Podobnie jak w komórkach nerwowych, nagromadzenie żelaza w enterocytach wierzchołkowych było skorelowane z większą ekspresją ferrytyny, co stwierdzono metodami immunohistochemicznymi [10,32]. La Vaute i wsp. [32] wykazali ponadto wzrost gęstości DMT1 i ferroportyny odpowiednio na błonie wierzchołkowej i boczno-podstawnej enterocytów absorpcyjnych. Badania Galy’ego i wsp. [20] nie potwierdziły jednak tych obserwacji.
Niezależnie od różnic w obrazie metabolizmu, uzyskanych w badaniach na dwóch modelach myszy z nokautem genu Irp2 (których przyczyną, jak sugerują Galy i wsp. [20], może być m.in. odmienne podłoże genetyczne myszy użytych do badań), wydaje się, że brak IRP2 powoduje daleko bardziej głębokie naruszenie ogólnoustrojowej homeostazy żelaza niż brak IRP1. Niewątpliwie znamienną cechą niedoboru IRP2 jest rozregulowanie potranskrypcyjnego mechanizmu kontroli syntezy ferrytyny, co prowadzi do drastycznego wzrostu poziomu tego białka w tkankach (mózg, dwunastnica, wątroba) i w konsekwencji do ograniczenia biologicznej dostępności jonów żelaza. Wpływ tych zmian molekularnych na fizjologię metabolizmu żelaza wymaga dalszych badań, w czym mogą się okazać przydatne myszy z tzw. warunkowymi i selektywnymi tkankowo nokautami genów Irp1 i Irp2 [18,19].
WCZESNE ZAMIERANIE ZARODKÓW MYSZY Z PODWÓJNYM NOKAUTEM GENÓW IRP1 I IRP2
Niezbędność zarówno IRP1, jak i IRP2 do prawidłowego rozwoju myszy już na etapie wczesnego okresu życia płodowego wykazano w pracy opublikowanej przez Smitha i wsp. [57]. Na podstawie genotypowania płodów uzyskanych w wyniku krzyżowania ze sobą myszy będących podwójnymi heterozygotami (Irp1+/– Irp2+/–), wśród 6,5-dniowych i starszych płodów nie wykryto genotypu Irp1–/– Irp2–/–. Genotyp taki zidentyfikowano natomiast w około 20% blastocyst, które wykazywały jednak morfologiczne anomalie. Blastocysty te charakteryzowały się brązowym zabarwieniem, które pochodziło od jonów żelazowych odłożonych w cząsteczkach ferrytyny. Autorzy sugerują, że pod nieobecność dwóch potencjalnych represorów translacji ferrytyny dochodzi do nadmiernej, niekontrolowanej jej syntezy, a w konsekwencji do całkowitego ograniczenia biodostępności żelaza, co staje się bezpośrednią przyczyną zamierania zarodków. Podobne wyniki dotyczące zamieralności zarodków (między 3,5 i 9,5 dniem życia płodowego) uzyskano w badaniach na zarodkach z nokautem genu FtH, kodującym podjednostkę H ferrytyny, wykazującą aktywność ferroksydazy [15]. W przypadku nokautu tego genu letalny dla zarodków okazuje się nadmierny wzrost stężenia jonów żelaza, które pod nieobecność podjednostki H nie mogą być utleniane i włączane do składających się wyłącznie z podjednostek L homopolimerów ferrytyny, a tym samym nie ulegają detoksyfikacji.
Drastyczne naruszenie homeostazy żelaza zarówno w stronę jego niedoboru, jak i nadmiaru prowadzi do całkowitego zahamowania rozwoju zarodka, w wyniku czego nie dochodzi do jego implantacji.
PODSUMOWANIE
Wnioski płynące z badań na myszach KO IRP1 i KO IRP2 są ważnym elementem toczącej się obecnie dyskusji nad specyficzną rolą każdego z IRP w regulacji metabolizmu żelaza w odpowiedzi na różne czynniki patofizjologiczne [7,13,39,41,49]. Z dyskusji tej wyłania się obraz IRP2 jako białka dominującego w potranskrypcyjnej regulacji homeostazy żelaza w warunkach fizjologicznych oraz w odpowiedzi na wahania poziomu żelaza [39,49]. Należy zaznaczyć, że przypisanie IRP2 określenia białka dominującego nie oznacza wyłączności. Przekonują o tym wyniki badań na myszach o genotypie Irp2–/– Irp1+/–, u których obserwowano gwałtowne zaostrzenie procesów neurodegeneracyjnych [56], jak również objawów niedokrwistości [20,32] w porównaniu z myszami o genotypie Irp2–/– Irp1+/+. Jak wykazano ostatnio, IRP1 nie jest również białkiem niezbędnym do kontroli metabolizmu żelaza w warunkach stresu tlenowego, gdy dominującą reaktywną formą tlenu jest O2˙– [59]. Z kolei zmiany w metabolizmie żelaza zachodzące pod wpływem innej reaktywnej formy tlenu, jaką jest nadtlenek wodoru (H2O2), odbywają się poprzez wybiórczą aktywację IRP1 [41]. Podobnie sugeruje się, że IRP1 odgrywa dominującą rolę w regulacji metabolizmu żelaza przez reaktywne formy azotu [7]. Wydaje się, ze różnorodna i swoista wrażliwość białek IRP na różne czynniki biologiczne jest elementem regulacyjnej plastyczności systemu IRP/IRE, co ma istotne znaczenie w subtelnym dostosowaniu metabolizmu żelaza do zmiennych warunków patofizjologicznych.
PIŚMIENNICTWO
[1] Abboud S., Haile D.J.: A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem., 2000; 275: 19906-19912
[PubMed] [Full Text HTML] [Full Text PDF]
[2] Andrews N.C.: Iron metabolism: iron deficiency and iron overload. Annu. Rev. Genom. Hum. Genet., 2000; 1: 75-98
[PubMed]
[3] Andrews N.C.: Iron homeostasis: insights from genetics and animal models. Nat. Rev. Genet., 2000; 1: 208-217
[PubMed]
[4] Bartosz G.: Druga twarz tlenu. Wydawnictwo Naukowe PWN, Warszawa, 2003
[5] Beaumont C., Leneuve P., Devaux I., Scoazec J.Y., Berthier M., Loiseau M.N., Grandchamp B., Bonneau D.: Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinemia and cataract. Nat. Genet., 1995; 11: 444-446
[PubMed]
[6] Bourdon E., Kang D.K., Ghosh M.C., Drake S.K., Wey J., Levine R.L., Rouault T.A.: The role of endogenous heme synthesis and degradation domain cysteines in cellular iron-dependent degradation of IRP2. Blood Cells Mol. Dis., 2003; 31: 247-255
[PubMed]
[7] Bouton C., Drapier J.C.: Iron regulatory proteins as NO signal transducers. Sci. STKE, 2003; 2003 (182): pe17
[PubMed]
[8] Bouton C., Oliveira L., Drapier J.C.: Converse modulation of IRP1 and IRP2 by immunological stimuli in murine RAW 264.7 macrophages. J. Biol. Chem., 1998; 273: 9403-9408
[PubMed] [Full Text HTML] [Full Text PDF]
[9] Cairo G., Pietrangelo A.: Iron regulatory proteins in pathobiology. Biochem. J., 2000; 352: 241-250
[PubMed] [Full Text HTML] [Full Text PDF]
[10] Cooperman S.S., Meyron-Holtz E.G., Ollivierre-Wilson H., Ghosh M.C., McConnell J.P., Rouault T.A.: Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood, 2005; 106: 1084-1091
[PubMed] [Full Text HTML] [Full Text PDF]
[11] Cox T.M., Alexander G.J., Sarkany R.P.: Protoporphyria. Semin. Liver Dis., 1998; 18: 85-93
[PubMed]
[12] Dandekar T., Stripecke R., Gray N.K., Goosen B., Constable A., Johansson H.E., Hentze M.W.: Identification of a novel iron-responsive element in murine and human erythroid d
aminolevulinic acid synthase mRNA. EMBO J., 1990; 10: 1903-1909
[PubMed] [Full Text HTML] [Full Text PDF]
[13] Eisenstein R.S.: Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu. Rev. Nutr., 2000; 20: 627-662
[PubMed]
[14] Fenton H.J.H.: The oxidation of tartaric amid in presence of iron. J. Chem. Soc., 1894; 65: 899-910
[15] Ferreira C., Bucchini D., Martin M.E., Levi S., Arosio P., Grandchamp B., Beaumont C.: Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem., 2000; 275: 3021-3024
[PubMed] [Full Text HTML] [Full Text PDF]
[16] Fleming M.D., Trenor C.C. 3rd, Su M.A., Foernzler D., Beier D.R., Dietrich W.F., Andrews N.C.: Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat. Genet., 1997; 16: 383-386
[PubMed]
[17] Galy B., Ferring D., Benesova M., Benes V., Hentze M.W.: Targeted mutagenesis of the murine IRP1 and IRP2 genes reveals context-dependent RNA processing differences in vivo. RNA, 2004; 10: 1019-1025
[PubMed] [Full Text HTML] [Full Text PDF]
[18] Galy B., Ferring D., Hentze M.W.: Generation of conditional alleles of the murine Iron Regulatory Protein (IRP)-1 and -2 genes. Genesis, 2005; 43: 181-188
[PubMed]
[19] Galy B., Ferring D., Hentze M.W.: Function of the IRE/IRP system in the duodenum: tissue-specific inactivation of the iron regulatory protein (IRP0-1 and (IRP2)-2 genes in the mouse. First Congress of the International BioIron Society, 22-26 May 2005, Book of Abstracts, p. 3
[20] Galy B., Ferring D., Minana B., Bell O., Janser H.G., Muckenthaler M., Schumann K., Hentze M.W.: Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood, 2005; 106: 2580-2589
[PubMed] [Full Text HTML] [Full Text PDF]
[21] Gunshin H., Allerson C.R., Polycarpov-Schwarz M., Rofts A., Rogers J.T., Kishi F., Hentze M.W., Rouault T.A., Andrews N.C., Hediger M.A.: Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett., 2001; 509: 309-316
[PubMed]
[22] Guo B., Phillips J.D., Yu Y., Leibold E.A.: Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J. Biol. Chem., 1995; 270: 21645-21651
[PubMed] [Full Text HTML] [Full Text PDF]
[23] Guo B., Yu Y., Leibold E.A.: Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J. Biol. Chem., 1994; 269: 24252-24260
[PubMed] [Full Text PDF]
[24] Hanson E.S., Foot L.M., Leibold E.A.: Hypoxia post-translationally activates iron-regulatory protein 2. J. Biol. Chem., 1999; 274: 5047-5052
[PubMed] [Full Text HTML] [Full Text PDF]
[25] Hanson E.S., Rawlins M.L., Leibold E.A.: Oxygen and iron regulation of iron regulatory protein 2. J. Biol. Chem., 2003; 278: 40337-40342
[PubMed] [Full Text HTML] [Full Text PDF]
[26] Henderson B.R., Seiser C., Kühn L.C.: Characterization of a second RNA-binding protein in rodents with specificity for iron-responsive elements. J. Biol. Chem., 1993; 268: 27327-27334
[PubMed] [Full Text PDF]
[27] Iwai K., Klausner R.D., Rouault T.A.: Requirements for iron-regulated degradation of the RNA binding protein, iron regulatory protein 2. EMBO J., 1995; 14: 5350-5357
[PubMed] [Full Text HTML] [Full Text PDF]
[28] Knutson M., Wessling-Resnick M.: Iron metabolism in the reticuloendothelial system. Crit. Rev. Biochem. Mol. Biol., 2003; 38: 61-88
[PubMed] [Full Text PDF]
[29] Kohler S.A, Henderson B.R, Kühn L.C.: SDHb mRNA of Drosophila melanogaster has a functional iron-responsive element in its 5′-untranslated region. J. Biol. Chem., 1995; 270: 30781-30786
[PubMed] [Full Text HTML] [Full Text PDF]
[30] Kohler S.A, Menotti E., Kühn L.C.: Molecular cloning of mouse glycolate oxidase. High evolutionary conservation and presence of an iron-responsive element-like sequence in the mRNA. J. Biol. Chem., 1999; 274: 2401-2407
[PubMed] [Full Text HTML] [Full Text PDF]
[31] Labbe R.F., Dewanji A.: Iron assessment tests: transferrin receptor vis-a-vis zinc protoporphyrin.. Clin. Biochem., 2004; 37: 165-174
[PubMed]
[32] La Vaute T., Smith S., Cooperman S., Iwai K., Land W., Meyron-Holtz E., Drake S.K., Miller G., Abu-Asab M., Tsokos M., Switzer R. 3rd, Grinberg A., Love P., Tresser N., Rouault T.A.: Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet., 2001; 27: 209-214
[PubMed] [Full Text HTML] [Full Text PDF]
[33] Levi S., Girelli D., Perrone F., Pasti M., Beaumont C., Corrocher R., Albertini A., Arosio P.: Analysis of ferritins in lymphoblastoid cell lines and in the lens of subjects with hereditary hyperferritinemia-cataract syndrome. Blood, 1998; 91: 4180-4187
[PubMed] [Full Text HTML] [Full Text PDF]
[34] Lipiński P.: Zaburzenie homeostazy żelaza w limfoblastach L5178Y myszy we wczesnej fazie parakrynnego działania tlenku azotu. Pr. Mater. Zoot., 2005; 12: 1-98
[35] Lipiński P.: Regulacja wewnątrzkomórkowej homeostazy żelaza u ssaków. Pr. Mater. Zoot., 2000; 57: 131-144
[36] Lipiński P., Starzyński R.R.: Regulacja ogólnoustrojowej homeostazy żelaza przez hepcydynę. Post. Hig. Med. Dosw., 2004; 58: 65-73
[PubMed] [Full Text PDF]
[37] Maines M.D., Gibbs P.E.: 30 some years of heme oxygenase: from a „molecular wrecking ball” to a „mesmerizing” trigger of cellular events. Biochem. Biophys. Res. Commun., 2005; 338: 568-577
[PubMed]
[38] McKie A.T., Marciani P., Rolfs A., Brennan K., Wehr K., Barrow D., Miret S., Bomford A., Peters T.J., Ferzaneh F., Hediger M.A., Hentze M.W., Simpson R.J.: A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell, 2000; 5: 299-309
[PubMed]
[39] Meyron-Holtz E.G., Ghosh M.C., Iwai K., LaVaute T, Brazzolotto X., Berger U.V., Land W., Ollivierre-Wilson H., Grinberg A., Love P., Rouault T.A.: Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J., 2004; 23: 386-395
[PubMed] [Full Text HTML] [Full Text PDF]
[40] Meyron-Holtz E.G., Ghosh M.C., Rouault T.A.: Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science, 2004; 306: 2087-2090
[PubMed] [Full Text HTML] [Full Text PDF]
[41] Mueller S.: Iron regulatory protein 1 as a sensor of reactive oxygen species. Biofactors, 2005; 24: 171-181
[PubMed]
[42] Nemeth E., Rivera S., Gabayan V., Keller C., Taudorf S., Pedersen B.K., Ganz T.: IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Invest., 2004; 113: 1271-1276
[PubMed] [Full Text HTML] [Full Text PDF]
[43] Pawliuk R., Tighe R., Wise R.J., Mathews-Roth M.M., Leboulch P.: Prevention of murine erythropoietic protoporphyria-associated skin photosensitivity and liver disease by dermal and hepatic ferrochelatase. J. Invest. Dermatol., 2005; 124: 256-262
[PubMed] [Full Text HTML] [Full Text PDF]
[44] Nemeth E., Tuttle M.S., Powelson J., Vaughn M.B., Donovan A., Ward D.M., Ganz T., Kaplan J.: Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science, 2004; 306: 2090-2093
[PubMed] [Full Text HTML] [Full Text PDF]
[45] Picard V., Epsztejn S., Santambrogio P., Cabantchik Z.I., Beaumont C.: Role of ferritin in the control of the labile iron pool in murine erythroleukemia cells. J. Biol. Chem., 1998; 273: 15382-15386
[PubMed] [Full Text HTML] [Full Text PDF]
[46] Pierre J.L., Fontecave M.: Iron and activated oxygen species in biology: the basic chemistry. Biometals, 1999; 12: 195-199
[PubMed]
[47] Pietrangelo A.: Hereditary hemochromatosis – a new look at an old disease. N. Engl. J. Med., 2004; 350: 2383-2397
[PubMed]
[48] Ponka P., Lok C.N.: The transferrin receptor: role in health and disease. Int. J. Biochem. Cell Biol., 1999; 31: 1111-1137
[PubMed]
[49] Recalcati S., Alberghini A., Campanella A., Gianelli U., De Camilli E., Conte D., Cairo G.: Iron regulatory proteins 1 and 2 in human monocytes, macrophages and duodenum: expression and regulation in hereditary hemochromatosis and iron deficiency. Haematologica, 2006; 91: 303-310
[PubMed] [Full Text HTML] [Full Text PDF]
[50] Rothenberger, S., Müllner, E.W., Kühn, L.C.: The mRNA-binding protein which controls ferritin and transferrin receptor expression is conserved during evolution. Nucleic Acids Res., 1990; 18: 1175-1179
[PubMed] [Full Text HTML] [Full Text PDF]
[51] Roy C.N., Andrews N.C.: Anemia of inflammation: the hepcidin link. Curr. Opin. Hematol., 2005; 12: 107-111
[PubMed]
[52] Roy C.N., Enns C.A.: Iron homeostasis: new tales from the crypt. Blood, 2000; 96: 4020-4027
[PubMed] [Full Text HTML] [Full Text PDF]
[53] Sadlon T.J., Dell’Oso T., Surinya K.H., May B.K.: Regulation of erythroid 5-aminolevulinate synthase expression during erythropoiesis. Int. J. Biochem. Cell. Biol., 1999; 31: 1153-1167
[PubMed]
[54] Samaniego F., Chin J., Iwai K., Rouault T.A., Klausner R.D.: Molecular characterization of a second iron-responsive element binding protein, iron regulatory protein 2. Structure, function, and post-translational regulation. J. Biol. Chem., 1994; 269: 30904-30910
[PubMed] [Full Text PDF]
[55] Schalinske K.L., Blemings K.P., Steffen D.W., Chen O.S., Eisenstein R.S.: Iron regulatory protein 1 is not required for the modulation of ferritin and transferrin receptor expression by iron in a murine pro-B lymphocyte cell line. Proc. Natl. Acad. Sci. USA, 1997; 94: 10681-10686
[PubMed] [Full Text HTML] [Full Text PDF]
[56] Smith S.R., Cooperman S., La Vaute T., Tresser N., Ghosh M., Meyron-Holtz E., Land W., Ollivierre H., Jortner B., Switzer R. 3rd, Messing A., Rouault T.A.: Severity of neurodegeneration correlates with compromise of iron metabolism in mice with iron regulatory protein deficiencies. Ann. N.Y. Acad. Sci., 2004; 1012: 65-83
[PubMed]
[57] Smith S.R., Ghosh M.C., Ollivierre-Wilson H., Hang Tong W., Rouault T.A.: Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyst stage of embryonic development. Blood Cells Mol. Dis., 2006; 36: 283-287
[PubMed]
[58] Starzyński R.R., Lipiński P.: IRP1, białko kontrolujące homeostazę żelaza komórkach ssaków: regulacja jego aktywności przez jony żelaza tlenek azotu. Post. Biol. Kom., 2003; 30: 497-514
[Abstract]
[59] Starzyński R.R., Lipiński P., Drapier J.C., Diet A., Smuda E., Bartłomiejczyk T., Gralak M.A., Kruszewski M.: Down-regulation of iron regulatory protein 1 activities and expression in superoxide dismutase 1 knockout mice is not associated with alterations in iron metabolism. J. Biol. Chem., 2005; 280: 4207-4212
[PubMed] [Full Text HTML] [Full Text PDF]
[60] Venti A., Giordano T, Eder P, Bush A.I., Lahiri D.K., Greig N.H., Rogers J.T.: The integrated role of desferrioxamine and phenserine targeted to an iron-responsive element in the APP-mRNA 5′-Untranslated Region. Ann. N.Y. Acad. Sci., 2004; 1035: 34-48
[PubMed]
[61] Wang J., Chen G., Muckenthaler M., Galy B., Hentze M.W., Pantopoulos K.: Iron-mediated degradation of IRP2, an unexpected pathway involving a 2-oxoglutarate-dependent oxygenase activity. Mol. Cell. Biol., 2004; 24: 954-965
[PubMed] [Full Text HTML] [Full Text PDF]
[62] Weinberg E.D.: The lactobacillus anomaly: total iron abstinence. Persp. Biol. Med., 1997; 40: 578-583
[PubMed]
[63] Zheng L., Kennedy M.C., Blondin G.A., Beinert H., Zalkin H.: Binding of cytosolic aconitase to the iron responsive element of porcine mitochondrial aconitase mRNA. Arch. Biochem. Biophys., 1992; 299: 356-360
[PubMed]