
Wpływ mocy dawki na komórkowe, biochemiczne i molekularne efekty promieniowania jonizującego
Waldemar Przybyszewski 1 , Maria Wideł 2 , Agnieszka Szurko 3 , Zbigniew Maniakowski 4Streszczenie
Celem radioterapii jest letalne uszkodzenie komórki nowotworowej przy możliwym oszczędzeniu komórek prawidłowych. Końcowym efektem może być zarówno śmierć komórki w wyniku apoptozy lub nekrozy, jak i uszkodzenia cytogenetyczne prowadzące do śmierci komórek lub niestabilności genetycznej. Te destrukcyjne skutki promieniowania jonizującego są wynikiem m.in. nadutlenienia (peroksydacji) makrocząsteczek komórki, zwłaszcza bogatych w lipidy struktur błoniastych oraz lipidów chromatynowych, wywołanego przez bezpośrednią i pośrednią jonizację. Konsekwencją działania końcowych produktów peroksydacji są uszkodzenia DNA i białek. Cechą charakterystyczną popromiennych reakcji peroksydacji jest zjawisko efektu odwrotnie zależnego od mocy dawki promieniowania. Jest ono określane jako wzrost stopnia oksydacji, przy stałej dawce absorbowanej, towarzyszące obniżeniu mocy dawki. Jednak mała moc dawki może powodować akumulację komórek w radiowrażliwej fazie cyklu komórkowego, G2, ponieważ punkty kontrolne cyklu komórkowego nie są wrażliwe na dawki o małej mocy. Potencjalnie moc dawki promieniowania może mieć decydujący wpływ na skuteczność radioterapii jak i na intensywność działań niepożądanych zarówno w tkankach objętych polem napromieniowania, jak i ogólnoustrojowych. Znajomość efektów biologicznych zależnych od mocy dawki promieniowania, z jednej strony może poszerzyć pole manewru terapeutycznego w radioterapii, a z drugiej przyczynić się do ograniczenia powikłań popromiennych. Praca przedstawia efekty biologiczne zależne od mocy dawki promieniowania.
Słowa kluczowe:promieniowanie jonizujące • moc dawki • efekt odwrotnie zależny od mocy dawki • peroksydacja lipidów • uszkodzenia DNA • apoptoza • nekroza • moc dawki w radioterapii
Summary
The aim of radiation therapy is to kill tumor cells while minimizing damage to normal cells. The ultimate effect of radiation can be apoptotic or necrotic cell death as well as cytogenetic damage resulting in genetic instability and/or cell death. The destructive effects of radiation arise from direct and indirect ionization events leading to peroxidation of macromolecules, especially those present in lipid-rich membrane structures as well as chromatin lipids. Lipid peroxidative endproducts may damage DNA and proteins. A characteristic feature of radiation-induced peroxidation is an inverse dose-rate effect (IDRE), defined as an increase in the degree of oxidation (at constant absorbed dose) accompanying a lower dose rate. On the other hand, a low dose rate can lead to the accumulation of cells in G2, the radiosensitive phase of the cell cycle since cell cycle control points are not sensitive to low dose rates. Radiation dose rate may potentially be the main factor improving radiotherapy efficacy as well as affecting the intensity of normal tissue and whole-body side effects. A better understanding of dose rate-dependent biological effects may lead to improved therapeutic intervention and limit normal tissue reaction. The study reviews basic biological effects that depend on the dose rate of ionizing radiation.
Key words:ionizing radiation • dose rate • inverse dose rate • lipid peroxidation • DNA damage • apoptosis • necrosis • dose rate in radiotherapy
Moc dawki promieniowania jonizującego wydaje się stanowić ważny element radiowrażliwości. Obniżenie mocy przeważnie prowadzi do obniżenia efektu letalnego. Jednakże, w pewnych warunkach obserwuje się efekt odwrotny mocy dawki charakteryzujący się wzrostem uszkodzenia i śmiertelności komórek w porównaniu z efektem dawek równoważnych o dużej mocy [36,47]. Badania in vitro [27,78] i in vivo [20] wykazują, że zarówno promieniowanie X, γ, protony, jak również – ale w mniejszym stopniu – wysoko energetyczne cząstki o dużej liczbie atomowej Z (cząstki – HZE) powodują liniowy, zależny od dawki, wzrost oksydacji, cytotoksyczności, a także transformacji nowotworowej. Należy podkreślić, że różne rodzaje promieniowania jonizującego mogą w organizmach żywych powodować ilościowo podobne efekty, natomiast wydajność indukowania zmian biologicznych pozostaje zależna zarówno od liniowego transferu energii, LET, jak i rodzaju struktury biologicznej. Liniowy transfer energii jest miarą liczby jonizacji wywołanej przez promieniowanie w żywej tkance lub innym ośrodku, przypadającej na jednostkę odległości jaką przebywa promieniowanie. Promieniowanie o niskim współczynniku LET, poniżej 2 keV/µm, do którego zalicza się promieniowanie gamma, promieniowanie X, protony, powoduje słabą jonizację tkanek. Natomiast promieniowanie o wysokim współczynniku LET, powyżej 40 keV/µm, tj. cząstki α, wysokoenergetyczne cząstki HZE powodują wzmożoną jonizację. Teoretycznie, wzrost gęstości jonizacji mógłby sprzyjać obniżeniu radiotoksyczności wskutek zwiększonej częstotliwości zachodzenia reakcji rekombinacji powstających wolnych rodników. Wyniki badań doświadczalnych wykazały, że nasilona jonizacja w obrębie ważnych makromolekuł komórkowych, z powodu wyczerpania miejscowej puli antyoksydantów, powoduje rozległe uszkodzenia DNA niepodlegające naprawie (pęknięcia podwójnej nici DNA). Słaba jonizacja promieniowaniem o niskim współczynniku LET prowadzi do takich uszkodzeń DNA (pęknięcia pojedynczej nici DNA), które mogą być skutecznie naprawiane [37]. Uszkodzenia DNA nie są jednak wyłączną przyczyną toksycznego działania promieniowania, a znaczącą rolę w radiotoksyczności odgrywają produkty peroksydacji (nadutlenienia) ważnych dla życia makrocząsteczek komórki. Cechą charakterystyczną popromiennych procesów peroksydacji jest zjawisko efektu odwrotnie zależnego od mocy dawki promieniowania (inverse dose rate effect – IDRE). Jest ono definiowane jako wzrost stopnia peroksydacji, przy stałej dawce absorbowanej, towarzyszące obniżeniu mocy dawki promieniowania. Jednakże odwrócony efekt mocy dawki ujawnia się nie tylko na poziomie biochemicznym, lecz także na poziomie komórkowym i molekularnym, jako obniżenie przeżycia, wzrost uszkodzeń DNA, wzrost apoptozy. Efekt odwrotnie zależny od mocy dawki może stanowić rozróżnienie między bezpośrednim efektem aktywności wolnych rodników a pośrednim efektem aktywności produktów peroksydacji lipidów. Takie rozróżnienie można uzasadnić tym, że tylko efekt pośredni charakteryzuje się dynamiką odwrotnie zależną od mocy dawki promieniowania jonizującego [30,72]. Odróżnia się ono od prawidłowego efektu mocy dawki, który charakteryzuje się zwiększeniem efektu promieniowania ze wzrostem mocy dawki.
Pomijanie zjawiska efektu odwrotnie zależnego od mocy dawki promieniowania może prowadzić do niedoszacowania ryzyka związanego z terapeutycznym stosowaniem promieniowania jonizującego. Panuje pogląd, że w radioterapii klinicznej nie pojawia się efekt mocy dawki promieniowania w obszarze mocy dawek stosowanych w radioterapii wiązką promieniowania uzyskiwaną z aparatów rentgenowskich, źródeł kobaltowych 60Co czy liniowych akceleratorów cząstek. Taki pogląd wydaje się uzasadniony dla mocy dawek w przedziale 1–10 Gy/min [22]. Przedział taki obejmuje bowiem zakres pracy większości stosowanych obecnie aparatów do radioterapii nowotworów. Potencjalnie moc dawki promieniowania, czyli wielkość opisująca szybkość napromieniowania, może mieć zasadniczy wpływ na skuteczność i na intensywność działań niepożądanych radioterapii, zarówno w tkankach objętych polem napromieniowania, jak i ogólnoustrojowych.
Praca przedstawia przegląd dotychczas poznanych biologicznych efektów promieniowania jonizującego zarówno na poziomie komórkowym, jak i na poziomie organizmu zależnych od mocy dawki.
MOC DAWKI PROMIENIOWANIA A PEROKSYDACJA LIPIDÓW
Następstwem radioterapii pozostaje letalne uszkodzenie komórki wywołane bezpośrednio i pośrednio jonizacją składników komórkowych [58]. Wynikiem jonizacji jest pojawienie się struktur wolnorodnikowych (free radicals – FR), aktywnych form tlenu (reactive oxygen species – ROS), aktywnych form azotu (reactive nitrogen species – RNS) charakteryzujących się krótkim okresem półtrwania oraz rodników organicznych o długim okresie półtrwania powyżej 20 godzin w temperaturze pokojowej [31]. Powstałe na skutek jonizacji rodniki (tab.1) mogą wpływać na wewnątrzkomórkowe, metaboliczne reakcje oksydoredukcyjne zaburzając ich fizjologiczną dynamikę, co przyczynia się do aktywacji mechanizmów zaostrzających lub łagodzących toksyczne działanie promieniowania jonizującego [77]. Peroksydacja lipidów jest procesem samorozprzestrzeniającym się (self propagating process), którego wynikiem jest pojawienie się wielu cytotoksycznych produktów ubocznych charakteryzujących się wysoką dyfuzyjnością. Są nimi: wodoronadtlenki lipidów, aldehydy lipidów, endoperoksydy i epoksydy. Związki te stanowią element ryzyka mutacji, niestabilności genetycznej oraz pojawiania się nowotworów wtórnych [7,59]. Czynniki te prawdopodobnie mogą być mediatorami uszkodzeń nie tylko komórek bezpośrednio napromieniowanych, lecz także komórek sąsiadujących lub odległych od miejsca napromieniowania, zjawiska znanego pod nazwą efektu sąsiedztwa (bystander effect) [49,64]. Ekspozycja na dawkę promieniowania poprzez skutek jonizacji przyczynia się do wielokrotnego wzmożenia fizjologicznych reakcji oksydacji. System antyoksydacyjny komórki w trakcie napromieniowania zapobiega niektórym efektom popromiennym, ale może ulec wyczerpaniu w miejscach intensywnego powstawania wolnych rodników. Uszkodzenie popromienne dotyczy również ważnych makromolekuł komórkowych biorących udział w fizjologicznej syntezie substancji prooksydacyjnych. Dynamiczna modyfikacja fizjologicznego stanu stresu oksydacyjnego przez promieniowanie jonizujące może wpływać na utrwalanie efektów biologicznych napromieniowania w odległym czasie od chwili pochłonięcia dawki.
Tabela 1. Skala czasowa procesów indukowanych promieniowaniem jonizującym i ich następstw

Udokumentowano obecność fosfolipidów, w jądrze komórkowym, stanowiących część struktury chromatyny [2]. Wykazano, że fosfolipidy znajdują się w kompartmentach jądra, gdzie następuje synteza, dojrzewanie i transport rybonukleoprotein. Zmiany w ich poziomie są związane ze stopniem dynamiki replikacji DNA. Wyniki badań wykazały, ze lipidy polarne wyizolowane z jąder komórek wątroby znacząco łatwiej podlegały peroksydacji aniżeli lipidy obojętne dlatego też chromatyna może stanowić docelowy element strukturalny komórki podlegający oksydacyjnym modyfikacjom [9]. Jednak analiza frakcji chromatynowych jąder wątroby szczura ujawniła niski poziom wielonienasyconych kwasów tłuszczowych w transkrypcyjnie aktywnej chromatynie, w porównaniu do transkrypcyjnie nieaktywnej chromatyny [41]. Wyniki tych badań sugerują jednak możliwość bezpośredniego peroksydacyjnego uszkodzenia, in situ, genomu komórki. Procesy peroksydacji stanowią endogenne źródło związków mutagennych i teratogennych [58,59,61]. Wynikiem takich reakcji jest tlenowa degradacja tych związków tłuszczowych do krótkołańcuchowych produktów aldehydowych [1,58]. W oparciu o podobieństwa strukturalne, aktywne krótkołańcuchowe aldehydy podzielono na trzy duże grupy: 2-alkenale, 4-hydroksy-2-alkenale, ketoaldehydy. Pierwsza grupa związków, tj. 2-alkenale charakteryzuje się dużą aktywnością chemiczną determinowaną dwoma centrami reakcyjnymi. Druga grupa, do której zaliczany jest 4-hydroksy-2-nonenal (HNE), reprezentuje najbardziej znaczącą grupę aldehydowych produktów peroksydacji lipidów. Wśród wielu produktów peroksydacji lipidów 4-hydroksynoneal (4-HNE) stanowi ważny związek ze względu na jego udział w regulacji wzrostu komórek, różnicowaniu się komórek i apoptozie, modyfikacji szlaków sygnałowych i ekspresji genów [4]. Powstający w dużych ilościach w trakcie peroksydacji wielonienasyconych kwasów tłuszczowych omega 6 (kwas linolowy, kwas arachidonowy), 4-hydroksy-2-nonenal indukuje większość efektów cytopatologicznych stanowiących konsekwencję stresu oksydacyjnego [43,60]. Trzecią ważną grupą aldehydów są ketoaldehydy, do której należy 4-okso-2-nonenal (ONE). Związek ten jest produktem rozpadu kwasu 13-hydroperoksy-9,11-oktadekadienowego (13-HPODE), będącego pierwszorzędowym produktem oksydacji kwasu linolowego i pojawia się jako związek pośredni w reakcjach syntezy 4-hydroperoksy-2E-nonenalu (4-HPNE), będącego związkiem prekursorowym 4-hydroksy-2-nonenalu (4-HNE) [34]. Następstwem tlenowego rozkładu fosfolipidów i estrów cholesterolowych jest pojawienie zrębów aldehydowych (core aldehydes) utlenionych fosfolipidów oraz resztkowych struktur (backbones) estrów cholesterolowych. Wykazano, że końcowe produkty peroksydacji lipidów powstają znacznie wydajniej po napromieniowaniu błon biologicznych [29] oraz komórek [62,63] dawkami o małej mocy w porównaniu z działaniem dawek o dużej mocy (ryc. 1).
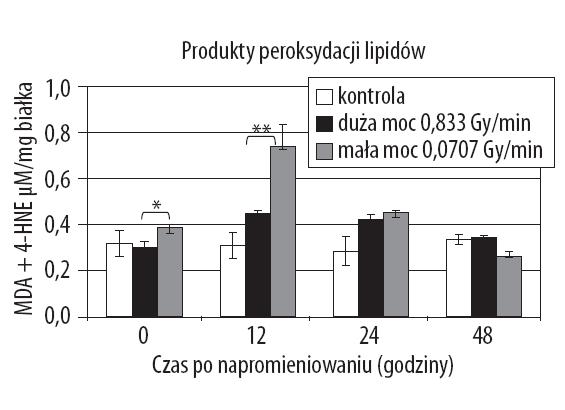
Ryc. 1. Poziom peroksydacji lipidów (MDA+4HNE) w komórkach linii AT478 po napromieniowaniu dawką 5 Gy, in vitro, o różnej mocy dawki. Duża moc dawki – 0,833 Gy/min. Mała moc dawki – 0,0707 Gy/min. Różnice znamienne statystycznie: * p=0,003, **p<0,003. Każdy punkt czasowy przedstawia średnią ± odchylenie standardowe z trzech hodowli i dwóch niezależnych eksperymentów; na podstawie [64] – zmodyfikowano
Badania biochemiczne skutków działania małej mocy dawki promieniowania X ujawniły, że strukturalna modyfikacja białek błonowych i cytosolowych w obecności dodanych lipidów pochodzenia błonowego i liposomów zbudowanych z nienasyconych lipidów była następstwem popromiennego uszkodzenia. Jednocześnie w badanych liposomach z dodatkiem witaminy E lub nasyconych kwasów tłuszczowych nie obserwowano strukturalnych modyfikacji białek w zależności od mocy dawki promieniowania [30]. Również efekt inaktywacji błonowego enzymu Na/K-ATP-azy był znacząco większy po dawce promieniowania o małej mocy [23]. Głównym chemicznym następstwem uszkodzenia DNA przez utlenione lipidy są pęknięcia pojedynczej i podwójnej nici DNA (SSBs i DSBs) [81]. Zaobserwowano, że hydroksyalkenale w stężeniach mikromolarnych powodowały w komórkach ludzkiej białaczki fragmentację nukleosomalnego DNA. Proces ten jest wynikiem aktywności kaspaz aktywujących DN-azę degradującą DNA (caspase activated deoxyribonuclease – CAD) [82].
USZKODZENIA DNA A MOC DAWKI PROMIENIOWANIA
Promieniowanie jonizujące powoduje wiele różnorodnych uszkodzeń DNA i białek, a także i innych makromolekuł komórkowych stanowiących sygnały, które aktywują białkowe sensory stymulujące zatrzymanie cyklu komórkowego oraz procesy naprawcze [12]. Wyniki badań własnych [78] wykazały, że napromieniowanie in vivo mysich guzów LLC (Lewis lung carcinoma) promieniowaniem gamma 60Co w dawkach 2–6 Gy o mocy 1 Gy/min lub 0,34 Gy/min indukowało w komórkach izolowanych z tych guzów i inkubowanych in vitro znacznie wyższy poziom uszkodzeń DNA w postaci mikrojąder po zastosowaniu dawek o małej mocy (ryc. 2).

Ryc. 2. Zależny od mocy dawki odsetek mikrojąder oceniany w komórkach mysiego raka płuc (LLC) wyizolowanych z guzów napromieniowanych in vivo dawkami 0–6 Gy; na podstawie [79] – zmodyfikowano
Również w naszych doświadczeniach napromieniowanie in vitro komórek raka mysiego linii AT478 dawką 5 Gy o małej mocy 0,071 Gy/min indukowało większe uszkodzenie DNA (ryc. 3) w porównaniu z działaniem takiej samej dawki o dużej mocy 0,83 Gy/min [63]. Zastosowanie testu kometowego (elektroforeza pojedynczych komórek na szkiełkach mikroskopowych) do analizy uszkodzeń i naprawy DNA in vitro w komórkach mięśniakomięsaka R1 i ludzkiego czerniaka linii Me45 napromieniowanych dawką 5 Gy fotonów X o mocy 0,83 i 0,071 Gy/min wykazało wprawdzie porównywalny poziom indukowanych uszkodzeń DNA po zastosowaniu dużej i małej mocy dawki, natomiast uszkodzenia indukowane dawką o małej mocy charakteryzowały się znacząco wolniejszą dynamiką naprawy aniżeli uszkodzenia wywołane dawką o dużej mocy [44]. Wolniejsza dynamika naprawy wynikała, prawdopodobnie, z małej aktywacji białkowych detektorów uszkodzeń DNA. Wykazano bowiem, że uszkodzenie DNA w komórkach raka okrężnicy i prostaty oraz prawidłowych fibroblastach spowodowane ekspozycją in vitro na dawkę 2 Gy o małej mocy, indukującą ~4–5 pęknięć DSBs/h w porównaniu z tą samą dawką o dużej mocy, indukującej ~1800 DSBs/h nie aktywuje kinazy białkowej ATM (ataxia teleangiectasia mutated) (EC 2.7.1.11) stanowiącej „czujnik” uszkodzenia DNA, oraz białek histonu H2AX i NBS1 (Nijmegen breakage syndrome1). Białko H2AX jest fosforylowane przez kinazę ATM w miejscu Ser-139, natomiast białko NBS1 w miejscu Ser-343 [8]. Ufosforylowane białko H2AX otacza uszkodzony fragment DNA, co stanowi sygnał do aktywacji czynników naprawy. Białko NBS1 natomiast wchodzi w skład kompleksu naprawczego MRE11-RAD51-NBS1 [75]. Napromieniowanie dawką o małej mocy powodowało obniżenie poziomu fosforylacji białka H2AX. Mała aktywacja ATM nie wzbudza zatem szlaku reperacji DNA zależnego od ATM, co powoduje wzrost śmiertelności komórek [10,79]. Wyniki opublikowanych badań wykazują jednak, że nie każda dawka o małej mocy powoduje wzrost śmiertelności komórek i obniżoną aktywację ATM i H2AX, co dowodzi istnienia wartości progowej uszkodzenia DNA i następowej aktywacji kinazy ATM [10].

Ryc. 3. Kinetyka naprawy pęknięć DNA w komórkach linii AT478 po napromieniowaniu dawką 5 Gy, in vitro, o różnej mocy dawki. Duża moc dawki – 0,833 Gy/min. Mała moc dawki – 0,0707 Gy/min. Różnice znamienne statystycznie: *p<0,05, **p<0,001, ***p<0,005. Każdy punkt czasowy przedstawia średnią ± odchylenie standardowe z trzech hodowli i dwóch niezależnych eksperymentów, na podstawie [64] – zmodyfikowano
POPROMIENNE ZMIANY W CYKLU KOMÓRKOWYM A MOC DAWKI I WIELKOŚĆ DAWKI PROMIENIOWANIA
Zarówno prawidłowe, jak i nowotworowe komórki reagują odmiennie na napromieniowanie dawkami o dużej lub małej mocy [39,73,77]. Zaobserwowano, że w komórkach proliferujących po napromieniowaniu dawką o dużej mocy aktywowane zostają punkty kontrolne cyklu komórkowego (cell cycle checkpoints) G1/S i G2/M, a zwłaszcza punkt G1/S, określający rodzaj naprawy uszkodzeń popromiennych lub początek procesu apoptozy. Wzrost apoptozy po napromieniowaniu małą mocą dawki obserwowano w komórkach szczurzego mięśniakomięsaka linii R1 [62] oraz komórkach HeLa Hep2 [46]. Mimo że wyniki badań na komórkach raka płaskonabłonkowego linii AT478 nie wykazały istotnych różnic w częstości apoptozy po napromieniowaniu dużą (0,833 Gy/min) i małą mocą (0,0707 Gy/min), to zaobserwowano wzrost poziomu nekrozy w komórkach napromieniowanych małą mocą dawki. Wydaje się, że wzrost nekrozy komórek może odzwierciedlać nasilenie uszkodzeń struktur błoniastych komórki oprócz niezreperowanych uszkodzeń DNA, wyrażających się w postaci mikrojąder [63]. Obserwowane różnice w odpowiedzi na moc dawki mogą sugerować odmienny stan obrony antyoksydacyjnej w różnych liniach komórkowych. Wyniki badań porównawczych in vitro efektów działania dużej i małej mocy dawki na aktywność głównych enzymów antyoksydacyjnych, dysmutazy ponadtlenkowej (MnSOD) i peroksydazy glutationowej (GSH-Pox) w komórkach raka płaskonabłonkowego linii AT478 ujawniły większy wzrost aktywności enzymów po dawce promieniowania o małej mocy jako wyraz wzmożonych procesów antyoksydacyjnych [63]. Wydłużony czas ekspozycji w trakcie napromieniowania dawką o małej mocy powoduje wzrost śmiertelności komórek w znacznie większym stopniu aniżeli krótka ekspozycja na dawkę promieniowania o dużej mocy. Mała moc dawki promieniowania raczej nie powoduje zatrzymania proliferacji komórek w punkcie kontrolnym G1/S, nie hamując przejścia komórek do dalszych faz cyklu komórkowego. Konsekwencją wydłużonego czasu napromieniowania może być gromadzenie komórek w radiowrażliwym punkcie kontrolnym G2/M [6]. Wyniki badań in vitro wpływu dawki promieniowania o małej mocy na cykl komórkowy różnych linii komórkowych raka stercza dowiodły, że popromienne zakłócenie replikacyjnego cyklu komórkowego dawką o małej mocy nie było powodem zwiększonej śmiertelności komórek [14]. Pozostaje zatem niewyjaśniona przyczyna efektu letalnego indukowanego przez dawki promieniowania o małej mocy. Badania radiowrażliwości asynchronicznych i synchronicznych hodowli utrwalonej linii ludzkich komórek (NHIK3025) wywodzących się z raka szyjki macicy (cervical carcinoma) in situ ujawniły większą wydajność inaktywacji komórek dawką o małej mocy w porównaniu z dawką o dużej mocy 60Co. Efekt taki, zaobserwowany dla dawek przekraczających wartość 7 Gy, prawdopodobnie był spowodowany premitotyczną akumulacją komórek w trakcie napromieniowania dawką o małej mocy [19]. Napromieniowanie ludzkich komórek raka sutka linii T-47D z funkcjonalnym genem RB (retinoblastoma) promieniowaniem gamma 60Co o różnej mocy dawki nie powodowało wzmożonej inaktywacji komórek przez dawkę o małej mocy w porównaniu z dawką o dużej mocy. Natomiast zaobserwowano, że około 15% populacji komórek T-47D zatrzymanych napromieniowaniem w fazie G2 cyklu komórkowego charakteryzowało się podwyższonym poziomem białka pRB będącego produktem genu RB [18]. Prawdopodobnie białko to w komórkach T-47D może być wskaźnikiem stresu oraz moduluje dynamikę cyklu komórkowego w warunkach stresu oksydacyjnego. Białko pRb jest substratem dla cyklinozależnych kinaz CDK2/4, których aktywność jest stymulowana przez cykliny D i A, a tłumiona przez białko p21. Zahamowanie proliferacji komórek ujawnia się brakiem fosforylacji białka pRb, które wiążąc się z czynnikiem transkrypcyjnym E2F inaktywuje go. Analogiczny efekt zaniku fosforylacji białka pRB obserwuje się w trakcie inkubacji zarówno komórek białaczki HL-60 jak i nerwiaka (neuroblastoma) linii SK-NBE z 4-hydroksynonenalem, co skutkowało zatrzymaniem proliferacji [5]. Pobudzenie proliferacji komórek wywołuje wzmożoną fosforylacją białka pRb, co prowadzi do wzrostu transkrypcyjnej aktywności czynnika E2F. Czynnik transkrypcyjny E2F wiąże się z promotorami wielu genów: c-myc, cdc2, cykliny A, c-myb, p21, których produkty warunkują progresję cyklu komórkowego.
Badania ostatnich kilku lat wykazały, że niekiedy po zastosowaniu bardzo małych dawek o dużej mocy występuje zjawisko przypominające odwrócony efekt mocy dawki. Po napromieniowaniu komórek dawkami poniżej 0,3 Gy o dużej mocy (~1 Gy/min) obserwuje się wzrost radiowrażliwości in vitro, co określone zostało mianem „radionadwrażliwości” na małe dawki promieniowania (low dose hyper-radiosensitivity, HRS). Komórki wykazujące radionadwrażliwość charakteryzują się mniejszą przeżywalnością po dawkach 0–0,5 Gy niż po dawce 1–2 Gy, co jest prawdopodobnie spowodowane mało wydajnie działającym radioochronnym mechanizmem naprawczym lub nie w pełni wykorzystaną zdolnością naprawy. Wzrost dawki promieniowania powoduje wzrost uszkodzeń DNA, co ułatwia mechanizmom naprawczym łatwiejsze rozpoznanie uszkodzenia, i w konsekwencji indukuje wzrost radiooporności (increased radioresistance – IRR) [42]. Radionadwrażliwość obserwowano również w przypadku frakcjonowania dawki promieniowania na ludzkiej linii komórek glejaka. Zastosowanie piętnastu frakcji 0,4 Gy powodowało większy efekt letalny aniżeli sześć frakcji 1,2 Gy [69]. Pozostaje do wyjaśnienia czy podobna radionadwrażliwość pojawiłaby się po zastosowaniu jeszcze mniejszych dawek stosowanych częściej i w liczniejszych frakcjach, a także po ciągłej ekspozycji małą mocą dawki. Wyniki doświadczeń in vitro napromieniowania dawką o małej mocy 60Co (poniżej 1 Gy/godz) ludzkich komórek raka stercza linii PC3 oraz ludzkich komórek glejaka (glioblastoma) linii T98G i A7, charakteryzujących się radionadwrażliwością na małą, pojedynczą dawkę o dużej mocy wykazały odwrócony efekt mocy dawki objawiający się większą śmiertelnością. Efekt taki nie wynikał z zatrzymania cyklu komórkowego w fazie G2/M, ale odzwierciedlał wrodzoną radionadwrażliwość, ponieważ jednocześnie badana linia komórek U373MG, niewykazująca cech radionadwrażliwości, nie wykazywała odwrotnie zależnego efektu mocy dawki [47]. Analiza faz cyklu komórkowego ludzkich komórek glejaka linii T98G i gwiaździaka (astrocytoma) linii U373 napromieniowanych jednorazowymi dawkami (0,05–5 Gy promieniowania X o mocy 0,2–0,4 Gy/min) ujawniła, że radionadwrażliwość (HRS) i następowa radiooporność (IRR) zaznaczały się w fazie G2 cyklu komórkowego [70]. Dla fazy G2 cyklu komórkowego są charakterystyczne dwa punkty kontrolne [80]. Pierwszy punkt kontrolny określany mianem „sinclair” aktywowany przez małe dawki promieniowania (0,3 Gy i poniżej) zatrzymuje w fazie G2 progresję cyklu komórek uszkodzonych promieniowaniem w fazie G1 lub S. Drugi punkt kontrolny fazy G2 nazywany „wczesnym” (early), aktywowany przez białko ATM i niewrażliwy na dawki promieniowania w zakresie 1–10 Gy, przeciwdziała dalszej progresji uszkodzonych przez promieniowanie komórek w fazie G2 [32,76]. Dlatego też zatrzymanie cyklu komórkowego we wczesnej fazie G2 (punkt kontrolny „sinclair”), pozwala na naprawę DNA komórek uszkodzonych promieniowaniem i wzrost przeżycia, a tym samym na przejście stanu radionadwrażliwości (HRS) w stan zwiększonej radiooporności (IRR). Wykazano ścisły związek między przemianą stanów HRS/IRR a pobudzeniem wczesnego punktu kontrolnego [42]. Jednak wykazanie analogicznego stopnia aktywacji kinazy ATM w liniach komórkowych niewykazujących radionadwrażliwości sugeruje, ze aktywacja ATM nie stanowi jednak zasadniczego elementu mechanizmu regulującego przeżywalność komórek [33]. Napromieniowanie hodowli niedzielących się fibroblastów ludzkich linii MRC5 i linii HSF1, HSF2 bardzo małymi dawkami promieniowania X (~1 mGy) wykazało, że pęknięcia podwójnej nici DNA (DSBs) pozostawały nienaprawione przez wiele dni w przeciwieństwie do szybkiej i wydajnej naprawy DSBs po dużych dawkach (200 mGy) [67]. Komórki napromieniowane małymi dawkami promieniowania, które przechodzą przez punkty kontrolne fazy G2 cyklu komórkowego z uszkodzeniami DNA nieaktywującymi kinazy białkowej ATM, osiągają stan mitozy, a następnie podlegają apoptozie [15]. Przemiana stanu HRS w stan IRR wydaje się zatem stanowić próg aktywacyjny mocy dawki promieniowania, zależny również od linii komórkowej [36]. Dane doświadczalne wskazują, że napromieniowanie dawką o małej mocy przed zastosowaniem małej pojedynczej dawki o wysokiej mocy może niwelować zjawisko HRS [47] z powodu wcześniejszej aktywacji białka ATM i procesów naprawy DNA [10]. Aktywację białka ATM uważa się za zasadniczy element składowy komórkowej odpowiedzi na ekspozycję małą mocą dawki [26,48]. Sugeruje się, że naprawa przez homologiczną rekombinację (homologous recombination – HR) jest podstawowym mechanizmem naprawczym w fazie G2 cyklu komórkowego [35]. Należy podkreślić, że homologiczna rekombinacja stanowi ważny mechanizm naprawczy dużych peroksydacyjnych uszkodzeń DNA [59]. Sugeruje się, że obserwowane zjawiska radionadwrażliwości (HRS) i radiooporności (IRR) odzwierciedlają procesy które uwalniają napromieniowane populacje komórkowe od uszkodzonych komórek pojawiających się po ekspozycji na bardzo niskie dawki promieniowania jonizującego oraz prawdopodobnie na promieniowanie środowiska [48].
AKTYWACJA CZYNNIKÓW TRANSKRYPCYJNYCH
Sugeruje się, że promieniowanie indukując zmiany w wewnątrzkomórkowym metabolizmie oksydoredukcyjnym może stanowić element inicjujący aktywację sygnałów transdukcyjnych, czynników transkrypcyjnych oraz ekspresji genów [71]. Efekty działania czynników genotoksycznych, w tym promieniowania jonizującego wykazują ścisły związek między pojawianiem się dwuniciowych pęknięć DNA (DSBs) a aktywacją czynników transkrypcyjnych [12]. Jak dotąd, wyniki badań wpływu napromieniowania na aktywację czynników transkrypcyjnych odnosiły się do działania dawek supraletalnych, które następnie podlegały ekstrapolacji na działania dawek małych, stosowanych klinicznie, tj. < Gy. Takie ekstrapolacje okazały się niewłaściwe zarówno dla większości znanych czynników transkrypcyjnych: p53, NFκB, AP-1, EGR-1, jak i indukowanych napromieniowaniem białkowych transkryptów. Dotychczas, w sposób odtwarzalny, udokumentowano aktywację trzech czynników transkrypcyjnych, a mianowicie: p53, NF-κB oraz SP1-(RCPs) po napromieniowaniu małymi dawkami promieniowania, które nie upośledzały przeżycia komórek [11]. W związku z tym rzeczywisty wpływ stosowania dawek klinicznych promieniowania na aktywację czynników transkrypcyjnych i białek pozostaje mało poznany, a prawie nieznany w przypadku stosowania dawek o małej mocy. Po napromieniowaniu komórek białaczki linii HL-60 dawką 20 Gy wykrywano transkrypty genu c-jun, we wszystkich fazach cyklu komórkowego, podobnie jak i nukleosomalną fragmentację DNA, co przemawiałoby za efektem koindukcji transkrypcji genu c-jun i fragmentacji DNA [40]. Dwudziestokrotne obniżenie mocy dawki promieniowania połączone było ze wzrostem poziomu transkryptów genu c-jun. Podobną zależność ekspresji tego genu od mocy dawki promieniowania obserwowano po jednorazowych dawkach promieniowania, tj.10 i 20 Gy [68]. Prawdopodobnie produkt genu c-jun wpływa na aktywność genów naprawy DNA, co skutkowało wzrostem przeżycia komórek po zastosowaniu dawki o małej mocy promieniowania jonizującego.
Wykazano jednak, że produkty peroksydacji lipidów, tj. aldehydy o krótkim łańcuchu węglowym zapobiegają aktywowaniu czynnika transkrypcyjnego NF-κB oraz ekspresji TNF wskutek zahamowania fosforylacji inhibitora NF-κB (IκB) i następowej proteolizy [51]. Podobnie, hamujący wpływ mikromolarnych stężeń 4-hydroksy-2-nonenalu (HNE) na aktywację NFkB, obserwowano in vitro w komórkach linii ludzkiego raka okrężnicy i raka płuca [25]. Inkubacja ludzkich komórek białaczkowych linii K562 i HL-60 z 4-HNE (1 µM) prowadziła do zahamowania ekspresji genu c-myc. Stopień zahamowania ekspresji był zależny od dawki i czasu inkubacji [4,5]. Jednak inkubacja hemopoetycznych komórek macierzystych (HSC) z bardzo niskimi (niecytotoksycznymi) dawkami 4-HNE lub 4-hydroksy-2,3-alkenali (10–6 M.) o różnej długości łańcucha indukowała m.in. wzrost transkrypcji i syntezę prokolagenu typu I, a także czynnika transkrypcyjnego AP-1. Podobne wyniki uzyskiwano w wyniku inkubacji komórek HSC z prooksydantami lub ROI (reactive oxygen intermediates). Hydroksyalkenale w zależności od wielkości dawki zastosowanej hamują proliferację komórek co jest bezpośrednio związane z ich cytotoksycznością [4,65].
EKSPRESJA GENÓW A MOC DAWKI NAPROMIENIOWANIA
Jak dotychczas, wyniki badań ekspresji genów w zależności od mocy dawki promieniowania jonizującego nie są spójne. Z jednej strony, napromieniowanie ludzkich fibroblastów skóry, in vitro, dawką małej mocy indukowało wzrost stężenia produktu białkowego genu GADD45A [13]. Z drugiej strony, analiza poziomu transkryptów genu interleukiny IL-1β i IL-6 w liniach komórkowych ludzkiego glejaka napromieniowanych in vitro dawkami o dużej i małej mocy wykazała, że dawka o małej mocy obniżała, natomiast dawka o dużej mocy stymulowała wzrost liczby transkryptów [66]. Wyniki badań poziomu mRNA genu HSP70 w komórkach płuc, śledziony i jelita cienkiego myszy napromieniowanych in vivo małą mocą dawki ujawniły znaczący wzrost jego ekspresji w analizowanych tkankach [45]. Kilkukrotne obniżenie mocy dawki promieniowania jonizującego przeciwdziałało apoptozie i indukowało liniowy wzrost poziomu produktu białkowego genów CDKN1A i GADD45A w ludzkich komórkach białaczkowych linii ML-1, in vitro, zachowujących funkcjonalny gen p53. Natomiast analiza poziomu produktu genu MDM2 nie wykazywała znaczących różnic w zależności od mocy dawki. Badanie ekspresji genów techniką mikromacierzową wykazało, że napromieniowanie ludzkich komórek białaczkowych radiowrażliwej linii ML-1 dawkami o różnej mocy indukuje dwie odmiennie reagujące grupy genów. Większość produktów białkowych genów znajdujących się w grupie o aktywności niezależnej od mocy dawki napromieniowania pełni rolę w regulacji proliferacji i progresji cyklu komórkowego, podobnie jak gen MDM2. Natomiast większość produktów białkowych genów znajdujących się w grupie aktywności zależnej od mocy dawki promieniowania charakteryzuje się znaczącym udziałem w procesie apoptozy, podobnie jak geny CDKN1A i GADD45A [3]. Mimo że związek między funkcją genu a efektem dawki promieniowania o różnej mocy nie jest absolutny, to sugeruje on jednak ilościowo różne mechanizmy odpowiedzi na małą i dużą moc dawki napromieniowania. Zwiększa to ryzyko kancerogenezy wskutek błędnej naprawy uszkodzenia popromiennego i kontynuowania proliferacji po napromieniowaniu dawkami o małej mocy. Produkty białkowe genów wrażliwych na moc dawki napromieniowania mogą stanowić użyteczny biologiczny marker ekspozycji na promieniowanie jonizujące.
KLINICZNE SKUTKI STOSOWANIA DAWEK O MAŁEJ MOCY I POTENCJALNE ZNACZENIE ZJAWISKA HRS
Klasyczna radioterapia frakcjonowana wykorzystuje promieniowanie jonizujące w zakresie dawek o dużej mocy, tj. od około 1 do kilku Gy/min [21], podczas gdy radioterapia śródtkankowa stosowana już w pierwszych latach po odkryciu radu opierała się na małych mocach dawek (około 0,5 Gy/godz) [53]. Już w 1941 r. Pierquin i Richard wykazali, że dawka 60 Gy podana w sposób ciągły w ciągu 5–6 dni była bardziej skuteczna w sterylizacji raka szyjki macicy niż ta sama dawka podana w sposób frakcjonowany w ciągu kilku tygodni. Bardzo trudno jest stosować napromieniowanie zewnętrzne promieniami o małej mocy ze względu na dyskomfort pacjenta poddawanego kilkugodzinnej sesji napromieniowania niezbędnej do dostarczenia dawki frakcyjnej 2 Gy lub wyższej, dlatego też kontrolowane doświadczenia kliniczne pochodzą z nielicznych ośrodków terapeutycznych, w tym z dwu ośrodków francuskich. W latach siedemdziesiątych ub.w. [54] zastosowano radioterapię LDR (low dose rate) (1 Gy/godz, 10 Gy/dzień) ze źródła 60Co w zaawansowanym raku gardła środkowego. Ze względu na silne odczyny śluzówki, wymagające zmniejszenia pola po dawce 45 Gy, w kolejnych badaniach zastosowano dawkę o mocy 15 mGy/min, prowadząc jednocześnie w innym ramieniu badawczym radioterapię standardową techniką frakcjonowania. Wyniki 3-letniej kontroli miejscowej (local control) wskazywały na większą skuteczność napromieniania LDR (84% wyleczeń miejscowych) nad standardową radioterapią (39% wyleczeń miejscowych), przy akceptowalnym nasileniu odczynów [55]. Kolejne badanie kliniczne przeprowadzono w grupie chorych na raka piersi w latach 1986-1989. Opublikowane niedawno wyniki 15-letniej obserwacji tych chorych [56] wykazują, że radioterapia dawkami o małej mocy (1,5 cGy/min, 5 sesji po 9 Gy) jest bardziej skuteczna, niż ta sama dawka w terapii konwencjonalnej, aczkolwiek występowanie nasilonych późnych odczynów popromiennych było częstsze w przypadku terapii LDR kiedy stosowano dawkę 45 Gy. Po obniżeniu dawki LDR do 35 Gy częstość odczynów była porównywalna z częstością odczynów po radioterapii konwencjonalnej. Natomiast stopień nasilenia odczynów popromiennych u chorych poddanych radioterapii LDR był porównywalny ze stopniem nasilenia odczynów u pacjentów poddanych konwencjonalnej radioterapii frakcjonowanej. Zaobserwowany efekt zarówno w postaci większej skuteczności terapii LDR w stosunku do guza, jak też większego odsetka odczynów był, jak sugerują autorzy, prawdopodobnie konsekwencją braku, lub tylko nieznacznej repopulacji guza podczas stosowania dawki o małej mocy, podczas gdy taka repopulacja występuje w czasie radioterapii frakcjonowanej. Ponieważ zdolność do repopulacji może różnić się między tkankami zdrowymi i nowotworowymi sugeruje się, że dalsze obniżanie mocy dawki mogłoby powodować wzrost tolerancji tkanek zdrowych [56]. Inną przyczyną obserwowanego efektu klinicznego odwrotnie zależnego od mocy dawki może być to, że dawki o dostatecznie małej mocy nie mogą indukować mechanizmów naprawczych w komórkach napromieniowanych, czego potwierdzeniem mogą być obserwacje, iż bardzo małe moce dawki (1 mGy/min) powodują uszkodzenia DNA bez aktywacji systemów naprawczych [10,76]. Nie bardzo jednak wiadomo jaka jest progowa moc dawki, poniżej której nie są aktywowane drogi sygnałowe prowadzące do naprawy uszkodzeń.
Skuteczność różnej mocy dawki promieniowania można oceniać także na podstawie wyników radioterapii nowotworów po zastosowaniu brachyterapii dawkami o dużej mocy (high dose rate brachytherapy – HDR) i małej mocy (low dose rate brachytherapy – LDR). Wyniki badań są jednak bardzo zróżnicowane. Retrospektywne badania skuteczności brachyterapii LDR i HDR w raku szyjki macicy nie wykazały znamiennych różnic zarówno pod względem wyleczenia miejscowego i przeżycia chorych, jak również późnych odczynów popromiennych [16]. Brachyterapia raka szyjki macicy, dawkami o małej i dużej mocy, której wyniki przedstawiają dwa niezależne opracowania pochodzące z Uniwersytetu Medycznego w Wisconsin [52] oraz brazylijskiego ośrodka onkologicznego w Sao Paulo [17] jednoznacznie wykazuje wyższą skuteczność małej mocy dawki, ale tylko w bardziej zaawansowanych stadiach nowotworu. Oceniono, że brachyterapia raka szyjki macicy dawkami o małej mocy (55 cGy/min), zastosowana po zakończeniu napromieniowania zewnętrznego, była bardziej skuteczna niż brachyterapia dawkami o dużej mocy, zarówno dla miejscowego wyleczenia guza (75 vs 44%, p=0,002), jak i całkowitego przeżycia chorych (58 vs 33%, p=0,004) w stopniu zaawansowania III B [52]. Nie obserwowano natomiast różnic w skuteczności brachyterapii LDR i HDR u chorych w przypadku niższych stopni zaawansowania, tj. I B i II B. Podobne wnioski wynikają z retrospektywnego opracowania [17] dotyczącego porównania skuteczności brachyterapii z dużą i małą mocą dawki w raku szyjki macicy. Tylko w przypadku chorych w III stopniu zaawansowania brachyterapia LDR była skuteczniejsza niż HDR, zarówno pod względem przeżycia bezobjawowego (46 vs 36%, p=0,04), jak i całkowitego (49 vs 37%, p=0,03). Jednakże, brachyterapia LDR powodowała wzrost powikłań ze strony odbytnicy (16%) w porównaniu z brachyterapią HDR (8%). Nie było natomiast znamiennych różnic w odsetku powikłań ze strony jelita cienkiego i pęcherza moczowego. Mniejsze prawdopodobieństwo powikłań ze strony odbytnicy po brachyterapii HDR jest, jak sądzą autorzy, związane z precyzyjną dozymetrią dawki w punkcie referencyjnym, czego nie można było przeprowadzić podczas brachyterapii LDR. Przeciwstawne do przedstawionych powyżej wyniki, uzyskano w brachyterapii LDR vs HDR gruczolakoraka szyjki macicy [57]. Wprawdzie 5-letnie przeżycie chorych było porównywalne w obu grupach, jednak odsetek powikłań był znamiennie wyższy po brachyterapii HDR (27%) niż po brachyterapii LDR (12%) [28]. Zwiększoną toksyczność brachyterapii HDR w porównaniu do LDR (p=0,005) odnotowano także w leczeniu raka pęcherza moczowego i autorzy konkludują, że brachyterapia HDR jest niewłaściwą metodą leczenia w przypadku tego nowotworu [57]. Wydaje się, że dotychczasowe badania nie dostarczają jednoznacznych wskazań do stosowania brachyterapii LDR w leczeniu nowotworów, a autorzy tych opracowań skłaniają się do konkluzji, że brachyterapia HDR jest metodą mniej uciążliwą dla pacjenta i bardziej bezpieczną dla personelu medycznego prowadzącego terapię.
Dyskusja nad potencjalnym klinicznym znaczeniem zjawiska radionadwrażliwości na małe dawki promieniowania jonizującego (HRS) skupia się na ocenie stopnia nasilenia efektów ubocznych radioterapii z zastosowaniem nowoczesnych technik napromieniania. Potencjalnie, pojawienie się HRS może w pewnych sytuacjach indukować wzrost efektu cytotoksycznego małych dawek w tkankach prawidłowych. Sugeruje się, że HRS może być wynikiem efektu sąsiedztwa (bystander effect), a zwiększona indywidualna wrażliwość na promieniowanie, związana z genetycznymi predyspozycjami może prawdopodobnie usposabiać do wystąpienia efektów w tkankach nienapromieniowanych dzięki temu zjawisku [49]. Wraz z rozwojem nowoczesnych technik, takich jak: trójwymiarowa radioterapia konformalna (3D conformal radiation therapy – 3D-CRT), i radioterapia intensywnie modulowaną dawką (intensity-modulated radiation therapy – IMRT), których celem jest obniżenie dawki w tkankach zdrowych, rośnie zagrożenie niepożądanymi efektami (m.in. ryzyko wtórnych nowotworów), wynikającymi ze stanu radionadwrażliwości i prawdopodobnego efektu sąsiedztwa, zwłaszcza, że przy tych technikach napromieniania większe objętości prawidłowych tkanek są eksponowane na mniejsze dawki. Efekt HRS zmniejszałby korzyści terapeutyczne wynikające z leczenia nowotworów intensywnie modulowaną dawką w porównaniu z terapią konwencjonalną [24,28]. Jednakże brak jest opracowań klinicznych dotyczących zjawiska radionadwrażliwości (HRS).
PIŚMIENNICTWO
[1] Agrawal A., Kale R.K.: Radiation induced peroxidative damage: mechanism and significance. Indian J. Exp. Biol., 2001; 39: 291-309
[PubMed]
[2] Albi E., Viola Magni M.P.: The role of intranuclear lipids. Biol. Cell., 2004; 96: 657-667
[PubMed]
[3] Amundson S.A., Lee R.A., Koch-Paiz C.A., Bittner M.L., Meltzer P., Trent J.M., Fornace A.J. Jr.: Differential responses of stress genes to low dose-rate γ irradiation. Mol. Cancer Res., 2003; 1: 445-452
[PubMed] [Full Text HTML] [Full Text PDF]
[4] Barrera G., Pizzimenti S., Dianzani M.U.: Lipid peroxidation: control of cell proliferation, cell differentiation and cell death. Mol. Aspects Med., 2008; 29: 1-8
[PubMed]
[5] Barrera G., Pizzimenti S., Laurora S., Briatore F., Toaldo C., Dianzani M.U.: 4-Hydroxynonenal and cell cycle. BioFactors, 2005; 24: 151-157
[PubMed]
[6] Bedford J.S., Mitchell J.B., Fox M.H.: Variation in responses of several mammalian cell lines to low dose-rate irradiation. In: Radiation Biology in Cancer Research. Ed: Meyn R.E., Withers R. New York: Raven Press 1980, pp.241-262
[7] Blair I.A.: DNA-adducts with lipid peroxidation products. J. Biol. Chem., 2008; 283: 15545-15549
[PubMed]
[8] Burma S., Chen B.P., Murphy M., Kurimasa A., Chen D.J.: ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem., 2001; 276: 42462-42467
[PubMed] [Full Text HTML] [Full Text PDF]
[9] Catala A., Cerruti A.: Non-enzymatic peroxidation of lipids isolated from rat liver microsomes, mitochondria and nuclei. Int. J. Biochem. Cell. Biol., 1997; 29: 541-546
[PubMed]
[10] Collis S.J., Schwaninger J.M., Ntambi A.J., Keller T.W., Nelson W.G., Dillehay L.E., DeWeese T L.; Evasion of early cellular response mechanisms following low level radiation-induced DNA damage. J. Biol. Chem., 2004; 279: 49624-49632
[PubMed] [Full Text HTML] [Full Text PDF]
[11] Criswell T., Leskov K., Miyamoto S., Luo G., Boothman D.A.: Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation. Oncogene, 2003; 22: 5813-5827
[PubMed] [Full Text HTML] [Full Text PDF]
[12] Davis T.W., Wilson-Van Patten C., Yang C.R., Sharada N., Meyers M., Kinsella T.J., Boothman D.A.: DNA repair in higher eukaryotes. W: DNA Damage and Repair, red.: Nickoloff J.A., Hoekstra M. Humana Press Inc. Totowa. NJ. 1998; t.2: 223-261
[13] DeToledo S.M., Azzam E.I., Gasmann M.K., Mitchel R.E.: Use of semiquantitative reverse transcription-plymerase chain reaction to study gene expression in normal human skin fibroblasts following low dose-rate irradiation. Int. J. Radiat. Biol., 1995; 67: 135-143
[PubMed]
[14] DeWeese T.L., Shipman J.M., Dillehay L.E., Nelson W.G.: Sensitivity of human prostatic carcinoma cell lines to low dose rate radiation exposure. J. Urol., 1998; 159: 591-598
[PubMed]
[15] Enns L., Bogen K.T., Wizniak J., Murtha A.D., Weinfeld M.: Low-dose radiation hypersensitivity is associated with p53-dependent apoptosis. Mol. Cancer Res., 2004; 2: 557-566
[PubMed] [Full Text HTML] [Full Text PDF]
[16] Falkenberg E., Kim R.Y., Meleth S., De Los Santos J., Spencer S.: Low-dose-rate vs. high dose-rate intracavitary brachytherapy for carcinoma of the cervix: The University of Alabama at Birmingham (UAB) experience. Brachytherapy, 2006; 5: 49-55
[PubMed]
[17] Ferrigno R., Nishimoto I.N., Dos Santos P.E., Pellizzon A.C., Maia M.A., Fogarolli R.C., Salvajoli J.V.: Comparison of low and high dose rate brachytherapy in the treatment of uterine cervix cancer. Retrospective analysis of two sequential series. Int. J. Radiat. Oncol. Biol. Phys., 2005; 62: 1108-1116
[PubMed]
[18] Furre T., Furre E.I., Koritzinsky M., Amellem O., Pettersen E.O.: Lack of inverse dose-rate effect and binding of the retinoblastoma gene product in the nucleus of human cancer T-47D cells arrested in G2 by ionizing radiation. Int. J. Radiat. Biol., 2003; 79: 413-422
[PubMed]
[19] Furre T., Koritzinsky M., Olsen D.R., Pettersen E.O.: Inverse dose-rate effect due to pre-mitotic accumulation during continuous low dose-rate irradiation of cervix carcinoma cells. Int. J. Radiat. Biol., 1999; 75: 669-707
[PubMed]
[20] Guan J., Wan X.S., Zhou Z., Ware J., Donahue J.J., Biaglow J.E., Kennedy A.R.: Effects of dietary supplements on space radiation-induced oxidative stress in Sprague-Dawley rats. Radiat. Res., 2004; 162: 572-579
[PubMed]
[21] Hall E.J., Brenner D.J.: The dose-rate effect revisited: radiobiological considerations of importance in radiotherapy. Int. J. Radiat. Oncol. Biol. Phys.,1991; 21: 1403-1414
[PubMed]
[22] Hall E.J.: Radiation dose-rate: a factor of importance in radiobiology and radiotherapy. Brit. J. Radiol., 1972; 45: 81-97
[PubMed]
[23] Hitschke K., Buhler R., Appel H.J., Stark G.: Inactivation of Na,K-ATPase by radiation-induced free radicals. Evidence for a radical-chain mechanisms. FEBS Lett., 1994; 353: 297-300
[PubMed]
[24] Honore H.B., Bentzen S.M.: A modeling study of the potential influence of low dose hypersensitivity on radiation treatment planning. Radiother. Oncol., 2006; 79: 115-121
[PubMed]
[25] Ji C., Kozak K.R., Marnett L.J.: IκB kinase, a molecular target for inhibition by 4-hydroxy-2-nonenal. J. Biol. Chem., 2001; 276: 18223-18228
[PubMed] [Full Text HTML] [Full Text PDF]
[26] Kato T.A., Nagasawa H., Weil M.M., Little J.B., Bedford J.S.: Levels of gamma-H2AX foci alter low-dose-rate irradiation reveal a DNA DSB rejoining defect in cells from human ATM heterozygotes in two AT families and in another apparently normal individual. Radiat. Res., 2006; 166: 443-453
[PubMed]
[27] Kennedy A.R., Ware J.H., Guan J., Donahue J.J., Biaglow J.E., Zhou Z., Stewart J., Vazquez M., Wan X.S.: Selenomethionine protects against adverse biological effects induced by space radiation. Free Radic. Biol. Med., 2004; 36: 259-266
[PubMed]
[28] Kim W.C., Kim G.E., Suh C.O., Loh J.J.: High versus low dose rate intracavitary irradiation for adenocarcinoma of the uterine cervix. Jpn. J. Clin. Oncol., 2001; 31: 432-437
[PubMed] [Full Text HTML] [Full Text PDF]
[29] Konings A.W.: Dose rate effects in biomembranes. J. Radiat. Res., 1979; 20: 259-263
[PubMed]
[30] Koufen P., Brdiczka D., Stark G.: Inverse-dose rate effects at the level of proteins observed in the presence of lipids. Int. J. Radiat. Biol., 2000; 76: 625-631
[PubMed]
[31] Koyama S., Kodama S., Suzuki K., Matsumoto T., Miyazaki T., Watanabe M.: Radiation-induced long-lived radicals which cause mutation and transformation. Mutat. Res., 1998; 421: 45-54
[PubMed]
[32] Krempler A., Deckbar D., Jeggo P.A., Lobrich M.: An imperfect G2M checkpoint contributes to chromosome instability following irradiation of S and G2 phase cells. Cell Cycle, 2007; 6: 1682-1686
[PubMed] [Full Text PDF]
[33] Krueger S.A., Collis S.J., Joiner M.C., Wilson G.D., Marples B.: The transition in survival from low-dose hyper-radiosensitivity to increased radioresistance is independent of activation of ATM SER1981 activity. Int. J. Radiat. Oncol. Biol. Phys., 2007; 69: 1262-1271
[PubMed]
[34] Lee S.H., Blair I.A.: Characterization of 4-oxo-2-nonenal as a novel product of lipid peroxidation. Chem. Res. Toxicol., 2000; 13: 698-702
[PubMed]
[35] Lehnert S., Chow T.Y.: Low doses of ionizing radiation induce nuclear activity in human tumour cell lines which catalyzes homologous double-strand recombination. Radiat. Environ. Biophys., 1997; 36: 67-70
[PubMed]
[36] Leonard B.E.: Tresholds and transitions for activation of cellular radioprotective mechanisms-correlations between HRS/IRR and the „inverse” dose rate effect. Int. J. Radiat. Biol., 2007; 83: 479-489
[PubMed]
[37] Limoli C.L., Ponnaiya B., Corcoran J.J., Giedzinski E., Kaplan M.I., Hartman A., Morgan W.F.: Genomic instability induced by high and low-LET ionizing radiation. Adv. Space Res., 2000; 25: 2107-2117
[PubMed]
[38] Lin P.S., Wu A.: Not all 2 Gray radiation prescription are equivalent: Cytotoxic effects depends on delivery sequences of partial fractionated doses. Int. J. Radiat. Oncol. Biol. Phys., 2005; 63: 536-544
[PubMed]
[39] Malaise E.P., Fertil B., Chavaudra N., Guichard M.: Distribution of radiation sensitivities for human tumor cells of specific histological types: comparison of in vitro and in vivo data. Int. J. Radiat. Oncol. Biol. Phys., 1986; 12: 617-624
[PubMed]
[40] Manome Y., Datta R., Taneja N., Shafman T., Bump E., Hass R., Weichselbaum R., Kufe D.: Coinduction of c-jun gene expression and internucleosomal DNA fragmentation by ionizing radiation. Biochemistry, 1993; 32: 10607-10613
[PubMed]
[41] Marmunti M., Catala A.: Non-enzymatic peroxidation of lipids isolated from rat liver nuclei and chromatin fractions. Int. J. Biochem. Cell. Biol., 1998; 30: 967-972
[PubMed]
[42] Marples B., Collis S.J.: Low-dose hyper-radiosensitivity: past, present, and future. Int. J. Radiat. Oncol. Biol. Phys., 2008; 70: 1310-1318
[PubMed]
[43] Matulewicz L., Przybyszewski W.M.: Wpływ lipidów na aktywność polimeraz DNA. Post. Biol. Kom., 2004; 31: 277-284
[Abstract]
[44] Matulewicz L., Wideł M.: Inverse dose rate effect on tumour cells measured by the comet assay. Pol. J. Environ. Studies, 2002; Suppl.II: 120-123
[45] Melkonyan H.S., Ushakova T.E., Umansky S.R.: HSP70 gene expression in mouse lung cells upon chronic γ-irradiation. Int. J. Radiat. Biol., 1995; 68: 277-280
[PubMed]
[46] Mirzaie-Joniani H., Eriksson D., Sheikholvaezin A., Johansson A.P., Lofroth P.O., Johansson L., Stigbrand T.: Apoptosis induced by low dose and low-dose-rate radiation. Cancer, 2002; 94 (4 Suppl.): 1210-1214
[PubMed] [Full Text HTML] [Full Text PDF]
[47] Mitchell C.R., Folkard M., Joiner M.C.: Effect of exposure to low-dose-rate 60Co gamma rays on human tumor cells in vitro. Radiat. Res., 2002; 158: 311-318
[PubMed]
[48] Mitchell C.R., Joiner M.C.: Effect of acute-dose irradiation on cell survival in vitro following low dose-rate exposures. Int. J. Radiat. Biol., 2002; 78: 981-990
[PubMed]
[49] Mothersill C., Moriarty M.J., Seymour C.B.: Radiotherapy and the potential exploitation of bystander effects. Int. J. Radiat. Oncol. Biol. Phys., 2004; 58: 575-579
[PubMed]
[50] Nakamura H., Yasui Y., Saito N., Tachibana A., Komatsu K., Ishizaki K.: DNA repair defect in AT cells and their hypersensitivity to low-dose-rate radiation. Radiat. Res., 2006; 165: 277-282
[PubMed]
[51] Page S., Fischer C., Baumgartner B., Haas M., Kreusel U., Loidl G., Hayn M., Ziegler-Heitbrock H.W., Neumeier D., Brand K.: 4-hydroxynonenal prevents NFκB activation and tumor necrosis factor expression by inhibiting IκB phosphorylation and subsequent proteolysis. J. Biol. Chem., 1999; 274: 11611-11618
[PubMed] [Full Text HTML] [Full Text PDF]
[52] Petereit D.G., Sarkaria J.N., Potter D.M., Schink J.C.: High- dose-rate versus low-dose-rate brachytherapy in the treatment of cervical cancer: Analysis of tumor recurrence – The University of Wisconsin Experience. Int. J. Radiat. Oncol. Biol. Phys., 1999; 45: 1267-1274
[PubMed]
[53] Pierquin B.: The optimization of delivered dose in radiotherapy: is it related to low dose rate? Radiother. Oncol., 2001; 58: 7-9
[PubMed]
[54] Pierquin B., Calitchi E., Mazeron J.J., LeBurgeois J.P., Leung S.: A comparison between low dose rate radiotherapy and conventionally fractionated irradiation in moderately extensive cancers of the oropharynx. Int. J. Radiat. Oncol. Biol. Phys., 1985; 11: 431-439
[PubMed]
[55] Pierquin B., Calitchi E., Mazeron J.J., LeBurgeois J.P., Leung S.: Update on low-dose rate irradiation for cancers of the oropharynx. Int. J. Radiat. Oncol. Biol. Phys., 1987; 13:259-261
[PubMed]
[56] Pierquin B., Tubiana M., Pan C., Lagrange J.L., Calitchi E., Otmezguine Y.: Long-term results of breast cancer irradiation treatment with low-dose-rate external irradiation. Int. J. Radiat. Oncol. Biol. Phys., 2007; 67: 117-121
[PubMed]
[57] Pos F.J., Horenblas S., Lebesque J., Moonen L., Schneider C, Sminia P, Bartelink H.: Low-dose-rate brachytherapy is superior to high-dose-rate brachytherapy for bladder cancer. Int. J. Radiat. Oncol. Biol. Phys., 2004; 59: 696-705
[PubMed]
[58] Przybyszewski W.M.: Udział produktów peroksydacji lipidów w przeciwnowotworowym mechanizmie działania promieniowania jonizującego i cytostatyków radiomimetycznych. Post. Hig. Med. Dośw., 2001; 55: 803-813
[PubMed]
[59] Przybyszewski W.M., Kasperczyk J., Stokłosa K., Bkhiyan A.: Uszkodzenia DNA powodowane przez produkty peroksydacji lipidów. Post. Hig. Med. Dośw., 2005; 59: 75-81
[PubMed] [Full Text HTML] [Full Text PDF]
[60] Przybyszewski W.M., Walichiewicz P.: Regulacja procesów wewnątrzkomórkowych przez kwasy tłuszczowe i końcowe produkty peroksydacji lipidów. Post. Hig. Med. Dośw., 2002; 56: 741-756
[PubMed]
[61] Przybyszewski W.M., Wideł M.: Kwasy tłuszczowe jak potencjalny element wspomagający w terapii nowotworów. Post. Hig. Med. Dośw., 2002; 56: 589-602
[PubMed]
[62] Przybyszewski W.M., Widel M., Palyvoda O.: Lipid peroxidation, DNA damage, and cellular morphology of rhabdomyosarcoma cell line irradiated in vitro by gamma-rays with different dose rate. Teratog. Carcinog. Mutagen., 2002; 22: 93-102
[PubMed]
[63] Przybyszewski W.M., Widel M., Polaniak R., Szurko A., Matulewicz L., Maniakowski Z., Birkner E., Rzeszowska-Wolny J.: Contrasting effects of low vs high dose-rate radiation on lipid peroxidation, DNA damage, and antioxidant enzyme activities in tumor cells. Progress in Medical Research, 2005; 3: 12-25
[64] Przybyszewski W.M., Wideł M., Szurko A., Lubecka B., Matulewicz Ł, Maniakowski Z., Polaniak R., Birkner E., Rzeszowska-Wolny J.: Multiple bystander effect on irradiated megacolonies of melanoma cells on non-irradiated neighbours. Cancer Lett., 2004; 214: 91-102
[PubMed]
[65] Robino G., Zamara E., Novo E., Dianzani M.U., Parola M.: 4-hydroxy-2,3-alkenals as signal molecules modulating proliferative and adaptative cell responses. BioFactors, 2001; 15: 103-106
[PubMed]
[66] Ross H.J., Canada A.L., Antoniono R.J., Redpath J.L.: High and low dose rate irradiation have opposing effects on cytokine gene expression in human glioblastoma cell lines. Eur. J. Cancer., 1977; 33: 144-152
[PubMed]
[67] Rothkamm K., Lobrich M.: Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl. Acad. Sci. USA, 2003; 100: 5057-5062
[PubMed] [Full Text HTML] [Full Text PDF]
[68] Sherman M.L., Datta R., Hallahan D.E., Weichselbaum R.R., Kufe D.W.: Ionizing radiation regulates expression of the c-jun protooncogene. Proc. Natl. Acad. Sci. USA, 1990; 87: 5663-5666
[PubMed] [Full Text HTML] [Full Text PDF]
[69] Short S.C., Kelly J., Mayes C.R., Woodcock M., Joiner M.C.: Low-dose hipersensitivity after fractionated low-dose irradiation in vitro. Int. J. Radiat. Biol., 2001; 77: 655-664
[PubMed]
[70] Short S.C., Woodcock M., Marples B., Joiner M.C.: Effects of cell phase on low-dose hyper-radiosensitivity. Int. J. Radiat. Biol., 2003; 79: 99-105
[PubMed]
[71] Spitz D.R., Azzam E.I., Li J.J., Gius D.: Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev., 2004; 23: 311-322
[PubMed]
[72] Stark G.: Functional consequences of oxidative membrane damage. J. Membr. Biol., 2005; 205: 1-16
[PubMed]
[73] Steel G.G.: Cellular sensitivity to low dose-rate irradiation focuses the problem of tumor radioresistance. Radiother. Oncol., 1991; 20: 71-83
[PubMed]
[74] Uchida K.: Lipid peroxidation and redox-sensitive signaling pathways. Curr. Atheroscler. Rep., 2007; 9: 216-221
[PubMed]
[75] Uziel T., Lerenthal Y., Moyal L., Andegeko Y., Mittelman L., Shiloh J.: Requirement of MRN complex for ATM activation by DNA damage. EMBO J., 2003; 22: 5612-5621
[PubMed] [Full Text HTML] [Full Text PDF]
[76] Vilenchik M.M., Knudson A.G.Jr.: Radiation dose-rate effects, endogenous DNA damage, and signaling resonance. Proc. Natl. Acad. Sci. USA, 2006; 103: 17874-17879
[PubMed] [Full Text HTML] [Full Text PDF]
[77] Wan X.S., Bloch P., Ware J.H., Zhou Z., Donahue J.J., Guan J., Stewart J., Kennedy A.R.: Detection of oxidative stress induced by low- and high-linear energy transfer radiation in cultured human epithelial cells. Radiat. Res., 2005; 163: 364-368
[PubMed]
[78] Widel M., Przybyszewski W.M.: Inverse dose-rate effect for the induction of micronuclei in Lewis Lung Carcinoma after exposure to cobalt-60 gamma-rays. Radiat. Res., 1998; 14: 98-102
[PubMed]
[79] Wykes S.M., Piasentin E., Joiner M.C., Wilson G.D., Marples B.: Low-dose hyper-radiosensitivity is not caused by a failure to recognize DNA double-strand breaks. Radiation Res., 2006; 165: 524
[PubMed]
[80] Xu B., Kim S.T., Lim D.S., Kastan M.B.: Two molecularly distinct G(2)/M checkpoints are induced by ionizing radiation. Mol. Cell. Biol., 2002; 22: 1049-1059
[PubMed] [Full Text HTML] [Full Text PDF]
[81] Yang M.H., Schaich K.M.: Factors affecting DNA damage caused by lipid hydroperoxides and aldehydes. Free Radic. Biol. Med., 1996; 20: 225-236
[PubMed]
[82] Zhang W., He Q., Chan L.L., Zhou F., El Naghy M., Thompson E.B., Ansari N.H.: Involvement of caspases in 4-hydroxy-alkenal-induced apoptosis in human leukemic cells. Free Radic. Biol. Med., 2001; 30: 699-706
[PubMed]